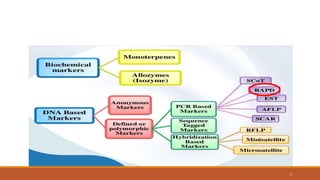
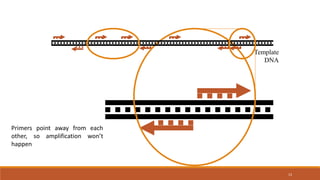
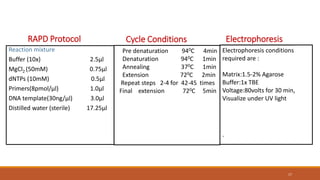
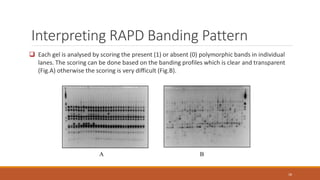
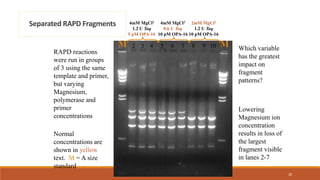
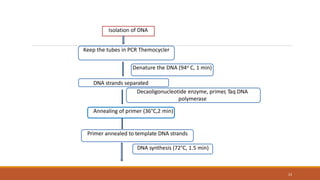
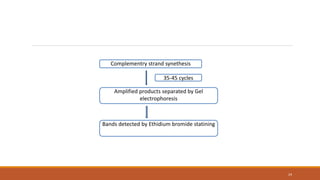
RAPD (Random Amplification of Polymorphic DNA) is a PCR-based molecular marker technique that involves using short, arbitrary nucleotide primers to randomly amplify genomic DNA fragments. These fragments can then be analyzed as genetic markers. RAPD works by using a single short primer to amplify random DNA sequences from a complex template. Variations in priming sites between individuals result in presence or absence of bands that can be used to analyze genetic relationships. The technique is fast, inexpensive and does not require prior DNA sequence knowledge, but results can lack reproducibility between laboratories.