Downloaded 92 times













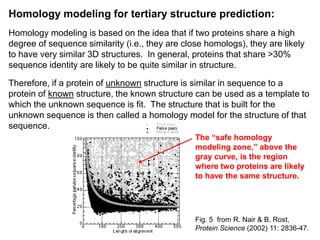



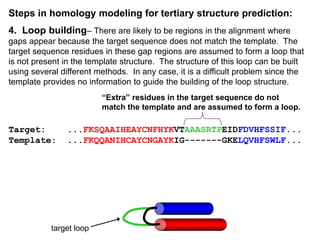







This document discusses protein structural bioinformatics and methods for predicting protein structure using bioinformatics approaches. It defines protein structural bioinformatics as focusing on representing, storing, analyzing and displaying protein structural information at the atomic scale. It describes how bioinformatics can be used to visualize, align, classify and predict protein structures. It also summarizes several specific methods for predicting protein secondary structure and tertiary structure, including homology modeling, threading and ab initio prediction.
















































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)












