Downloaded 16 times


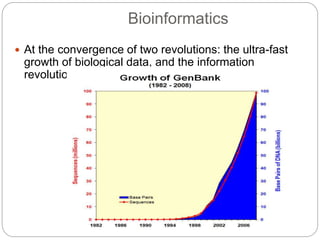
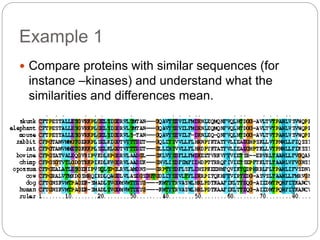
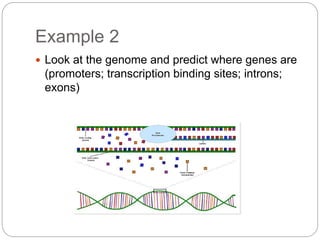
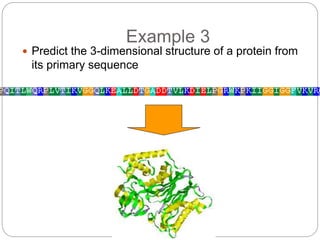
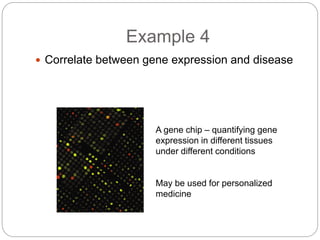










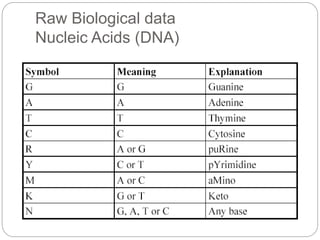
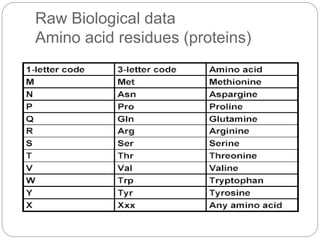
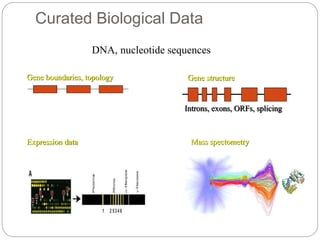
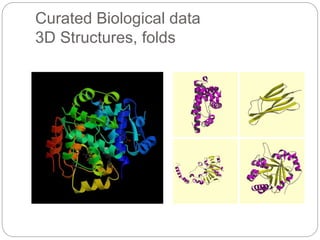
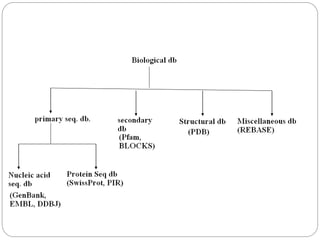
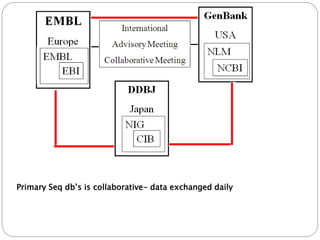








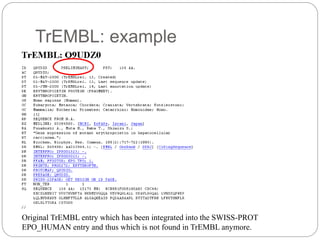






















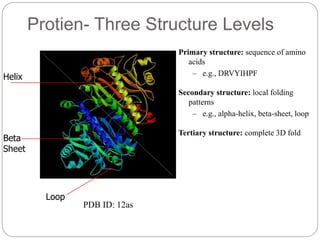
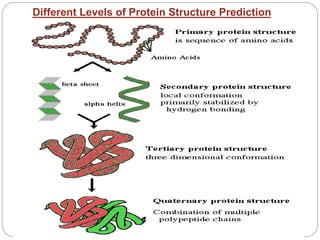
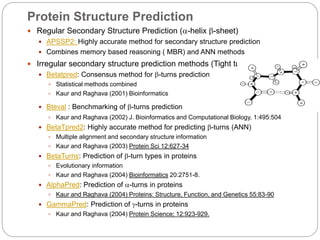
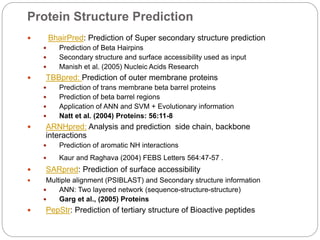
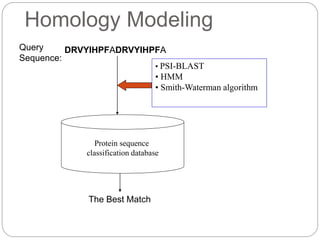

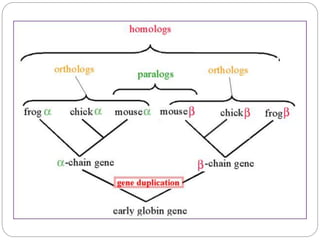
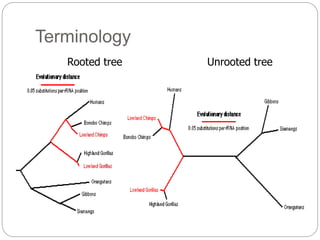

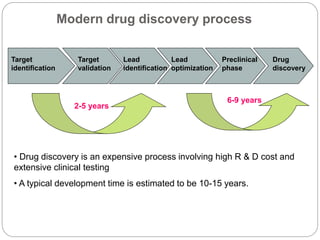

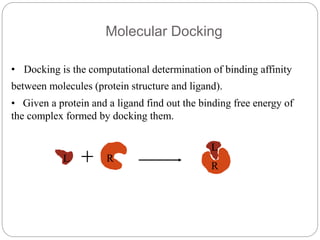



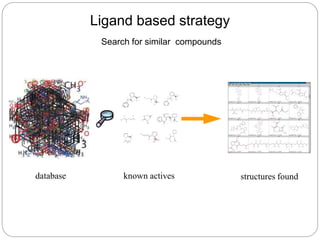

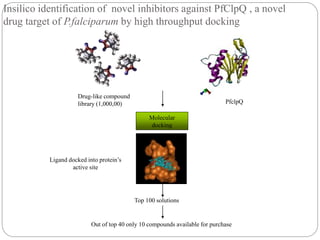



This document provides an overview of the field of bioinformatics. It discusses that bioinformatics is the analysis of biological information using computers and statistical techniques, and involves organizing, storing, analyzing and visualizing genomic data. It also discusses various databases used in bioinformatics, including nucleotide sequence databases like GenBank, protein sequence databases like Swiss-Prot, structure databases like PDB, and species-oriented databases. Examples of analyzing genomic sequences, predicting protein structures, and correlating gene expression and disease are also provided.









































































