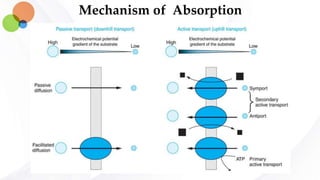
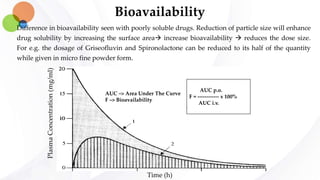
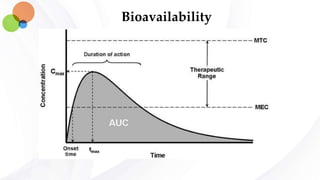
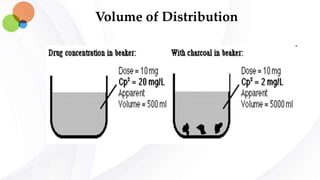
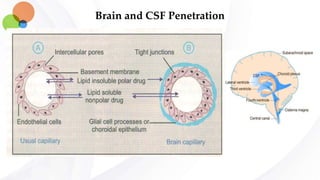
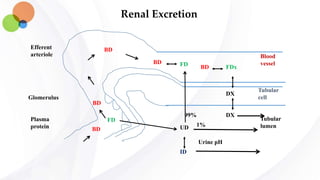
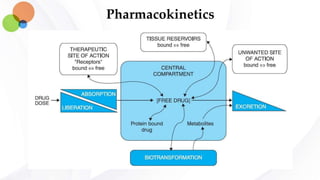
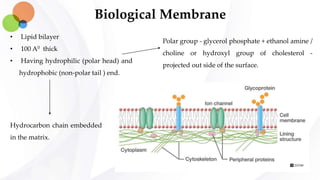
This document discusses pharmacokinetics and provides details about absorption, distribution, and bioavailability of drugs. It defines key pharmacokinetic terms and describes factors that influence absorption such as solubility, concentration, route of administration, and mechanisms of absorption including passive diffusion, active transport, and pinocytosis. Membrane permeability and drug properties like pH and lipid solubility are discussed. The document also covers volume of distribution, plasma protein binding, tissue storage, and barriers to drug distribution like the blood-brain barrier.




![Mechanism of Absorption
Drug absorption are influenced by pH:
Most of the drugs are weak electrolyte (weak acids or bases) and their ionization is pH dependent.
However, strong electrolyte completely dissociates. The ionization of weak electrolyte are given by
the equation:
pH = pKa + log
This equation relates the pH of the medium around the drug and the drug’s acid dissociation
constant (pKa) to the ratio of the protonated (HA or BH+) and unprotonated (A– or B) forms, where
HA→ A– + H+ (Ka = [A–][H+]/[HA]) describes the dissociation of an acid, and BH+ → B + H+,
( Ka = [B][H+]/[BH+]) describes the dissociation of the pronated form of a base.
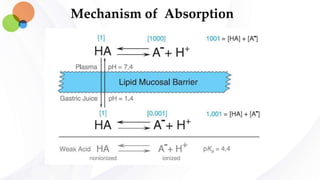
At steady state, an acidic drug will accumulate on the more basic side of the membrane and a basic
drug on the more acidic side—a phenomenon termed ion trapping.
[Protonated form]
[Unprotonated form]](https://image.slidesharecdn.com/pharmacokinetics-190418042147/85/Pharmacokinetics-7-320.jpg)