









































































































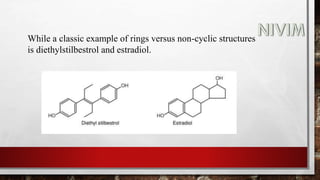





















![While simple metabolic reactions are followed by conjugation e.g., an
alkyl side chain of a drug may be oxidized to an alcohol which then
forms a conjugate with glucuronic acid or an ester may be hydrolysed to
its acid form which then, is coupled with glycine.
Examples:
[A] Oxidation:
Oxidation mainly occurs at aromatic rings, terminal positions of alkyl chains,
N-methyl groups, alcoholic groups and exposed corners of alicyclic rings.
(1) Ethanol, in mammals, is rapidly oxidised by liver alcohol dehydrogenases
to a toxic intermediate acetaldehyde. This is a reversible reaction.](https://image.slidesharecdn.com/unit-1-221129034207-c6ef41dc/85/Medicinal-Chemistrt-Unit-1-pptx-130-320.jpg)







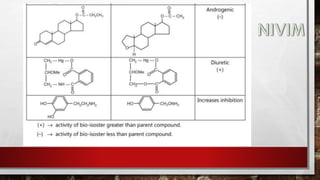
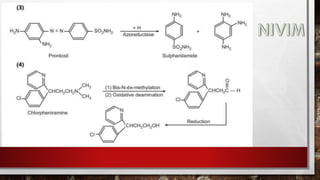
![[B] Reduction:
These processes play an important role in the metabolism of many
compounds containing carbonyl, nitro and azo groups.
Bioreduction of carbonyl compounds generates alcohol
derivatives, while nitro and azo reduction leads to amino
derivative. Since, the hydroxyl and amino groups are much more
susceptible to conjugation than the functional groups of the parent
compounds, reductive processes facilitate the drug elimination.](https://image.slidesharecdn.com/unit-1-221129034207-c6ef41dc/85/Medicinal-Chemistrt-Unit-1-pptx-140-320.jpg)

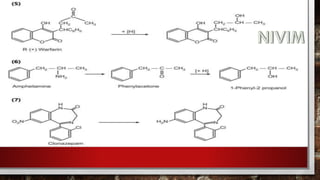
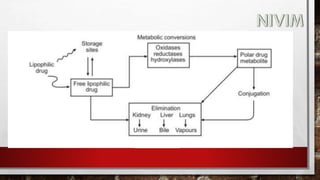
![[C] Hydrolytic Reactions:
The metabolism of ester and amide linkages in many drugs is
catalysed by hydrolytic enzymes present in liver, kidney, intestine,
blood and other tissues. The metabolic products formed, namely
carboxylic acids, alcohols, phenols and amines generally are polar
and functionally more susceptible to conjugation and excretion than
the parent ester and amides.
Amide hydrolysis appears to be mediated by liver microsomal
amidases, esterases and deacylases.](https://image.slidesharecdn.com/unit-1-221129034207-c6ef41dc/85/Medicinal-Chemistrt-Unit-1-pptx-144-320.jpg)
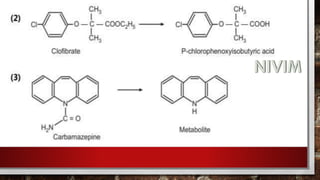

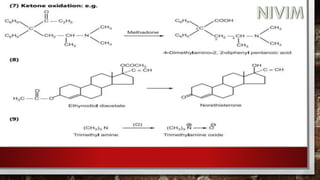
![[D] Other Important Reactions:
(1) N-dealkylation:](https://image.slidesharecdn.com/unit-1-221129034207-c6ef41dc/85/Medicinal-Chemistrt-Unit-1-pptx-147-320.jpg)





![(I) Conjugation with Glucuronic Acid:
The reaction involves the condensation of the drug or its metabolite
with the activated form of a readily available glucuronic acid, [i.e.
uridine diphosphate glucuronic acid (UDPGA)], which is synthesized
from glucose-1-phosphate. Glucuronic acid conjugation proceeds in
two steps.
(1) Formation of the activated form from glucose 1- phosphate.](https://image.slidesharecdn.com/unit-1-221129034207-c6ef41dc/85/Medicinal-Chemistrt-Unit-1-pptx-153-320.jpg)









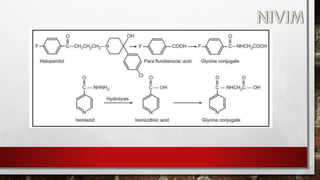


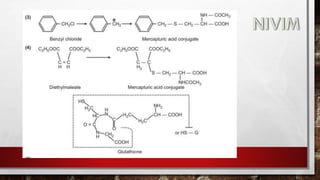
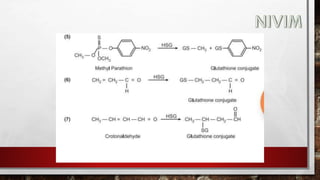































The document provides an extensive overview of medicinal chemistry, focusing on drug absorption, distribution, and the biological membrane's structure and functions. It discusses various factors influencing drug absorption, such as solubility and route of administration, and details the mechanisms by which drugs pass through membranes, including simple diffusion, active transport, and facilitated diffusion. It highlights the importance of understanding these processes to ensure drugs reach their intended sites of action effectively.























































































