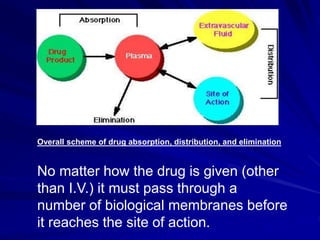
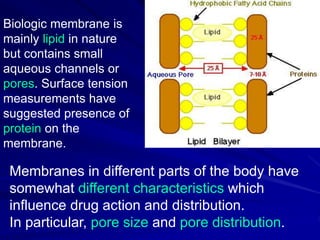
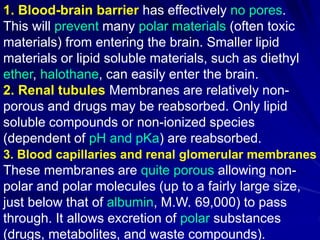
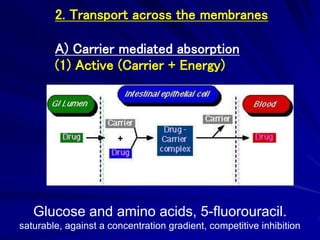
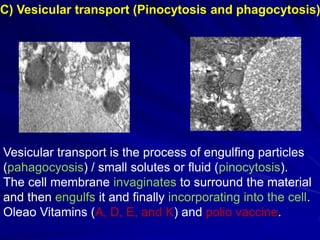
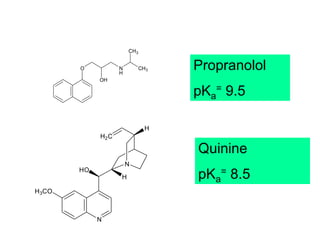
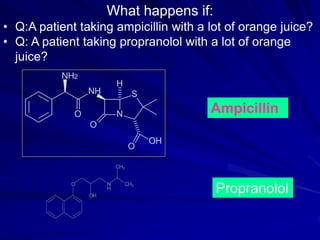
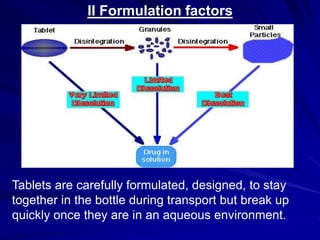
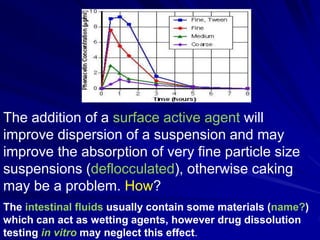
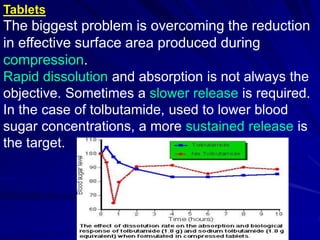
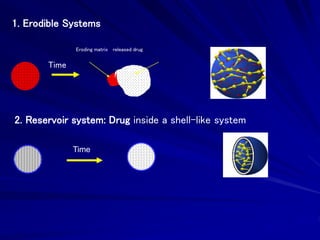
The document discusses biopharmaceutics, focusing on the physicochemical properties of drugs, drug delivery, and the pharmacokinetic processes affecting drug absorption, distribution, metabolism, and elimination. It elaborates on factors influencing drug bioavailability, including membrane physiology, transport mechanisms, and formulation considerations. The biopharmaceutics classification system (BCS) categorizes drugs based on solubility and permeability, aiding predictions of bioavailability and in vivo performance relationships.



























![Biopharmaceutics Classification System (BCS)
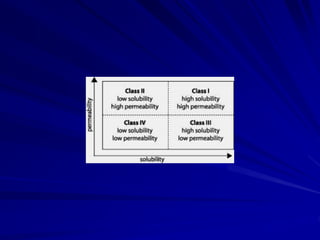
The biopharmaceutics classification system
(BCS) classifies drugs according to their
solubility and permeability. This enables
successful prediction of bioavailability from
solid oral dosage forms for drugs.
This classification system also provides a
guideline for determining the conditions under
which IVIVC is expected [Amidon et al, 1995].
For BCS class I drugs that have high solubility
and high permeability, the rate determining
step for drug absorption is likely to be drug
dissolution and gastric emptying rate.](https://image.slidesharecdn.com/biologicalandphysicochemicalfactors2017-2018-180208113221/85/Biological-and-physicochemical-factors-affecting-bioavailability-41-320.jpg)
![ The IVIVC for BCS class I drugs is expected if the
dissolution rate of the formulation is slower than
the gastric emptying rate. On the contrary, for
BCS class II drugs that have low solubility and
high permeability, the rate determining step for
drug absorption is the dissolution rate.
The IVIVC for BCS class II drugs is expected when
the in vitro drug dissolution rate is similar to the
in vivo drug dissolution rate [Dahan et al, 2009].
Whereas the BCS class III and class IV drugs
have poor permeability and the rate determining
step for drug absorption is most likely to be the
permeability across the GIT membrane. So the
IVIVC is not expected for BCS class III &class IV.](https://image.slidesharecdn.com/biologicalandphysicochemicalfactors2017-2018-180208113221/85/Biological-and-physicochemical-factors-affecting-bioavailability-42-320.jpg)




![Rate limiting steps in drug absorption [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/ratelimitingstepsindrugabsorptionautosaved-200623174235-thumbnail.jpg?width=640&height=640&fit=bounds)
























![Pharmacology - Pharmacokinetics 1 [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/pharmacology-pharmacokinetics1autosaved-241109192045-c128b252-thumbnail.jpg?width=640&height=640&fit=bounds)





































