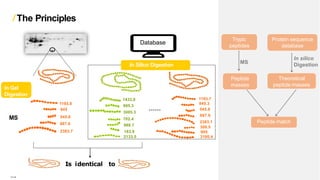
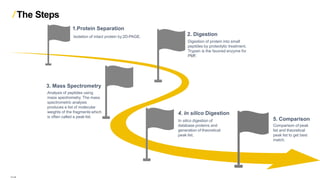
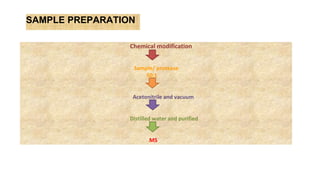
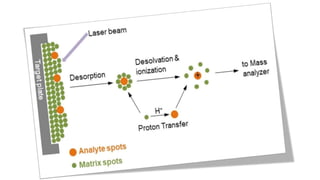
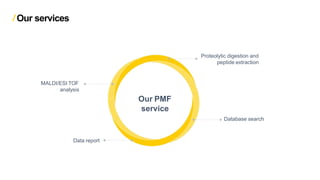
Peptide mass fingerprinting is a technique to identify proteins by breaking them into peptides via enzymatic digestion, measuring the peptide masses using mass spectrometry, and comparing the results to theoretical peptide masses from protein sequence databases to find a match. The key steps are isolating the protein, digesting it into peptides, using MALDI or ESI mass spectrometry to determine peptide masses, running an in silico digestion of protein databases to generate theoretical peptide masses, and comparing the experimental and theoretical masses to identify the protein.