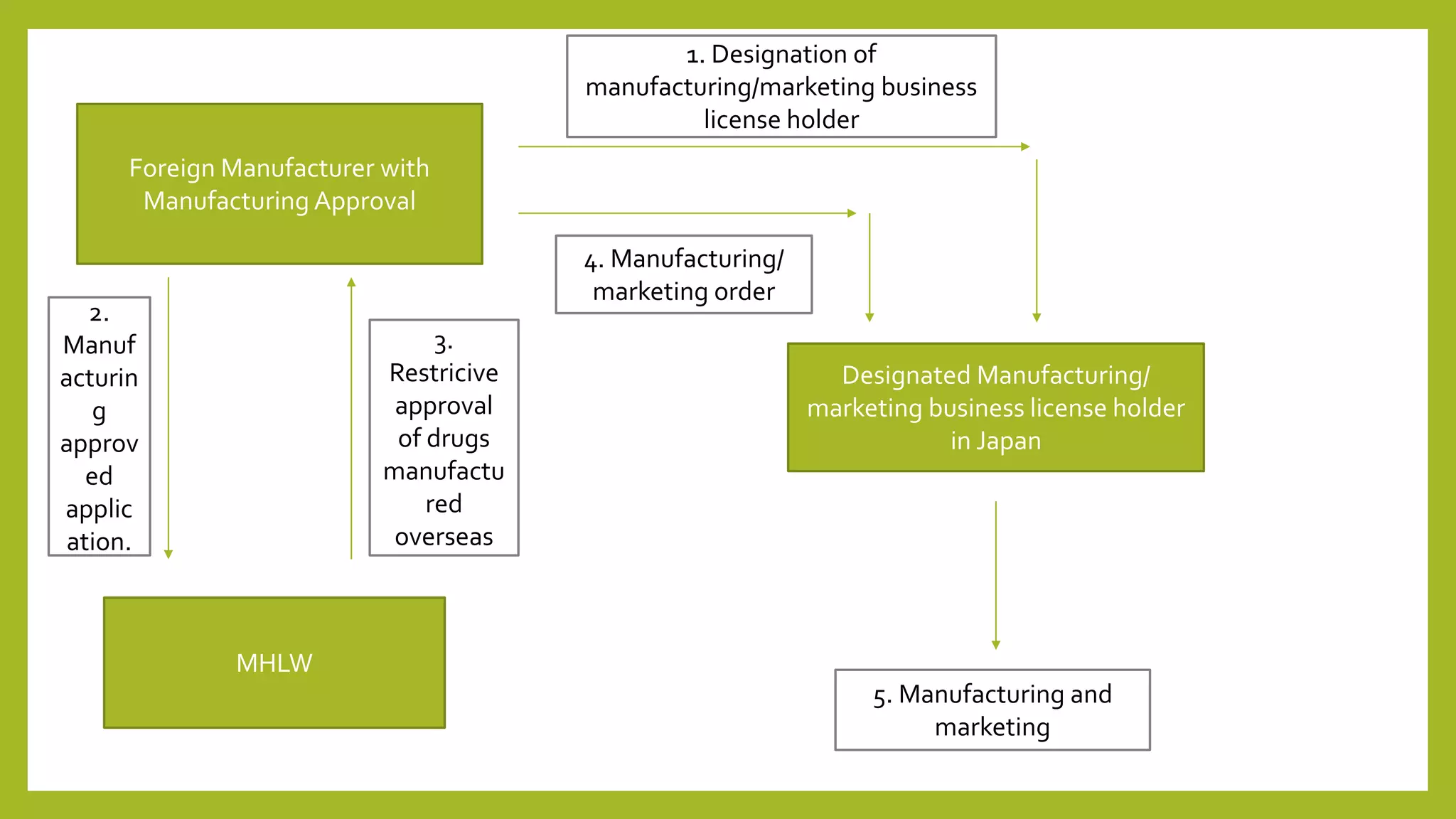
The document summarizes regulatory considerations for pharmaceuticals in Japan, including manufacturing, packaging, labeling, and post-marketing surveillance. For manufacturing, drugs must be approved by the Ministry of Health, Labor and Welfare and manufacturers must be licensed and follow good manufacturing practices. Packaging and labeling must contain specified information and any changes require relabeling. Post-marketing surveillance involves adverse event reporting, drug reexaminations every few years to reconfirm safety and efficacy, and reevaluations based on current medical knowledge.
























![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)





























































