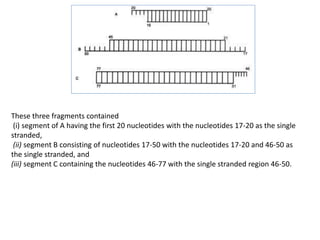
This document discusses gene synthesis techniques and blotting methods. It provides details on:
1) The first chemical synthesis of genes in the 1970s, including a gene for yeast tRNA and bacterial tRNA.
2) Methods for artificially synthesizing genes using oligonucleotides and ligating DNA fragments.
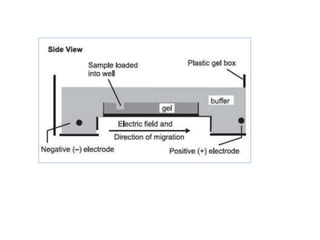
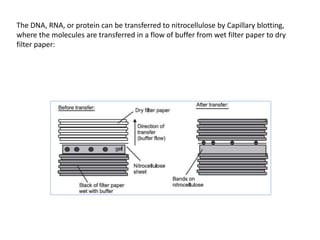
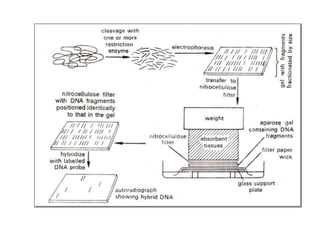
3) Techniques for analyzing DNA, RNA, and proteins - Southern blotting detects DNA, Northern blotting detects RNA, and Western blotting detects proteins.





























































![Elisa basics[2]](https://cdn.slidesharecdn.com/ss_thumbnails/elisabasics2-111218111650-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)











