Download as PDF, PPTX







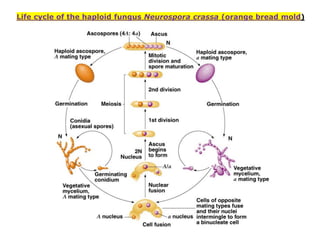

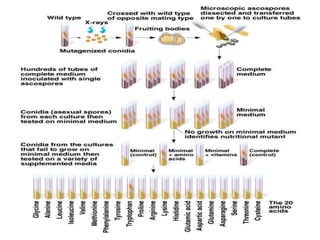
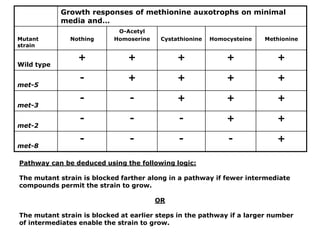
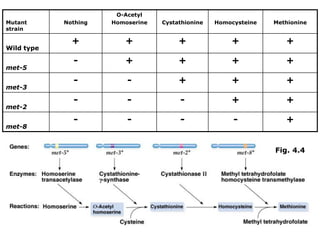

















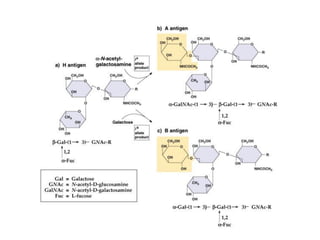




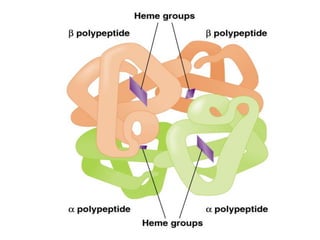






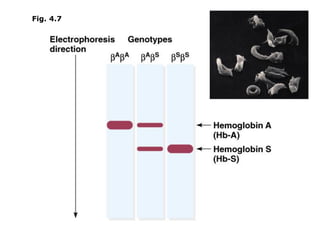

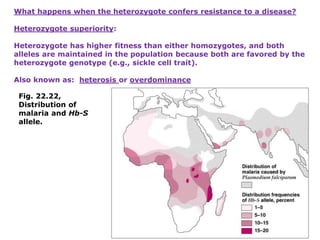


1. Beadle and Tatum's experiments with the fungus Neurospora crassa supported the "one gene-one enzyme hypothesis" which stated that each gene controls the production of a specific enzyme. 2. By inducing mutations in Neurospora and observing how they affected the fungus's ability to grow in different nutrient conditions, Beadle and Tatum were able to deduce biochemical pathways and determine that individual genes control steps in metabolism. 3. Their work provided evidence that genes control the synthesis of specific proteins and established the field of molecular genetics.























































































