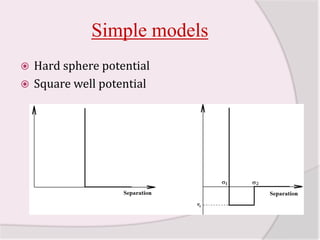
This document discusses molecular dynamics simulation methods. It begins by introducing molecular dynamics and Newton's laws of motion. It then describes how molecular dynamics simulations work by integrating Newton's equations of motion to obtain successive molecular configurations over time. Different algorithms for performing the integration are discussed, including the Verlet algorithm and leap-frog method. Predictor-corrector algorithms are also covered. The document provides guidance on best practices for setting up and running molecular dynamics simulations.