Download to read offline



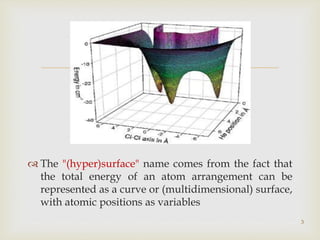
















The document discusses the conformation and potential energy of molecules, highlighting different methodologies for calculating these energies, including molecular mechanics and quantum mechanics. It emphasizes the role of molecular dynamics in simulating molecular interactions and the importance of energy minimization to identify local energy minima within a biomolecule's energy landscape. Additionally, it addresses the computational aspects and challenges associated with molecular dynamics simulations and the factors guiding molecular movement.










































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)







