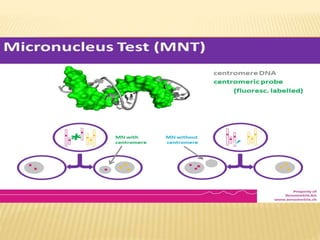
This document presents a procedure for conducting genotoxicity studies using in vitro and in vivo micronucleus assays. It introduces the micronucleus test as a method used in toxicological screening to identify potential genotoxic compounds. The procedure involves growing mammalian cells on cover glasses, treating the cells with test compounds, fixing and staining the cells, and examining them under a fluorescence microscope to detect micronuclei formation indicating DNA damage. References are provided for additional information on micronucleus testing methodology and applications.