Downloaded 60 times




























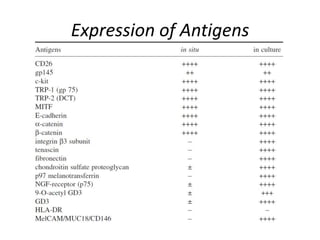


This document provides a detailed tutorial on the procedure for culturing human melanocytes. Key steps include: 1) Isolating epidermis from skin specimens using dispase enzyme solution overnight at 4°C. 2) Dispersing epidermal cells using trypsin and mechanically dissociating into a single cell suspension. 3) Seeding cells in TPA-free growth medium and incubating without disturbance for 48-72 hours. 4) Maintaining cultures by changing medium twice weekly and passaging confluent cultures using trypsin. Methods for cryopreserving and thawing melanocytes are also described. Morphology and growth characteristics of cultured melanocytes are provided.








































































































