
Downloaded 43 times







![EXPERIMENT
LOCALIZATION OF DISEASE GENE FOR INHERITED EYE
DISORDERS BY LINKAGE ANALYSIS
Introduction
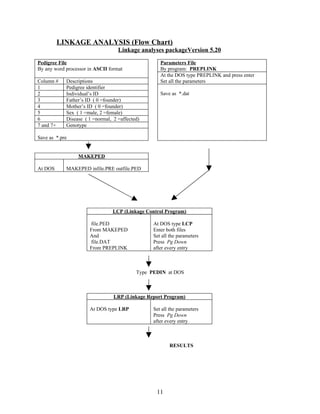
Linkage analysis is a powerful method of gene mapping. The primary goal is to determine if two
or more genetic traits – i.e. a marker locus (or multiple marker loci) and a disease trait – are co-
segregating within a pedigree. The results of linkage analyses are typically shown as “LOD
Scores” for a pair of loci at various recombination values. LOD stands for log to base 10 of
odds, and odds are that two loci are linked verses the two loci are not linked. LOD score is
calculated with help of computer programs: MLINK or LIPED which are part of the linkage
analyses package, as described in the Handbook of Human Genetic Linkage.
Equipment and Materials
10X PCR Buffer (Sigma) 100 mM Trizma-HCl, pH 8.3; 500 mM KCl; 15 mM MgCl2;
0.01% (w/v) gelatin
dNTPs 10 mM dATP; 10 mM dCTP; 10 mM dGTP; 10 mM
dTTP
Autoclaved deionized H2O aliquot in 1.5 ml eppendorf tubes
DNA samples 40 ng/ µl
Forward & Reverse Primers 20 µM
Taq Polymerase 2.5 U/µl
10X TBE Trizma base (Tris[hydroxymethyl]aminomethane) 70 g/L
boric acid 55g/L
EDTA (Ethylene diamine tetraacetic acid) 9.04 g/L
check pH (if not 8-8.2, then adjust the pH).
filter through millipore filter paper (0.45 µm).
40% Acrylamide solution Acrylamide 389.6g/L
N,N’-methylone bis-acrylamide 10.4g/L
store in dark bottles at 4oC
Gel loading dye 7.5% Ficoll
0.01% bromophenol blue
0.01% xylene cyanol
25% APS freshly prepared
TEMED
10% ethidium bromide
8](https://image.slidesharecdn.com/hgdpmanual-110804003901-phpapp02/85/Methods-in-molecular-Biology-8-320.jpg)


















This document describes an experiment to localize disease genes for inherited eye disorders through linkage analysis. It involves genotyping DNA samples from a pedigree using PCR and microsatellite markers. The results will be analyzed using linkage analysis software to calculate LOD scores and determine if marker loci are co-segregating with the disease trait, helping to map the location of the disease gene. Equipment and reagents needed for PCR, electrophoresis, and linkage analysis are listed.























































































