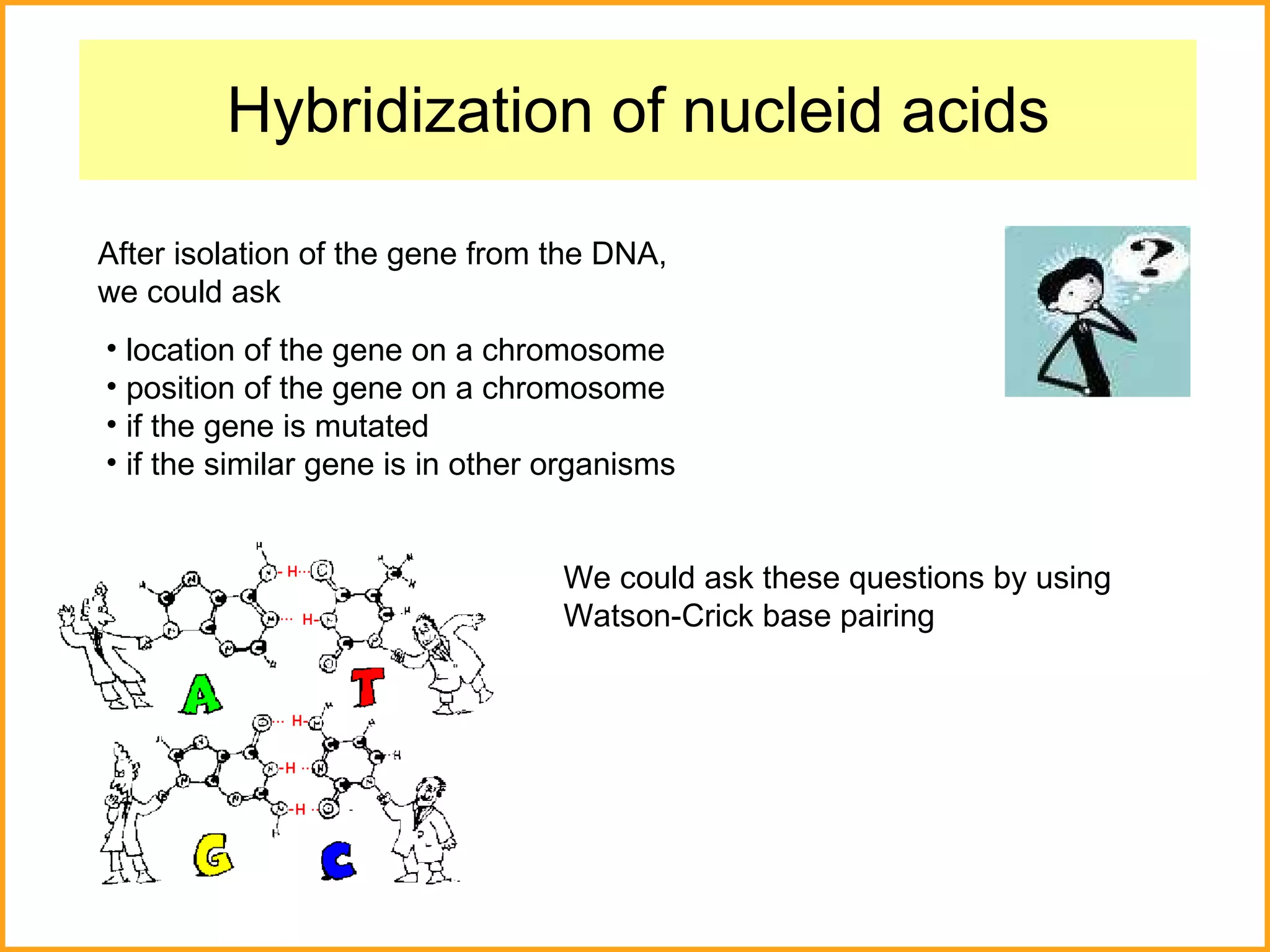
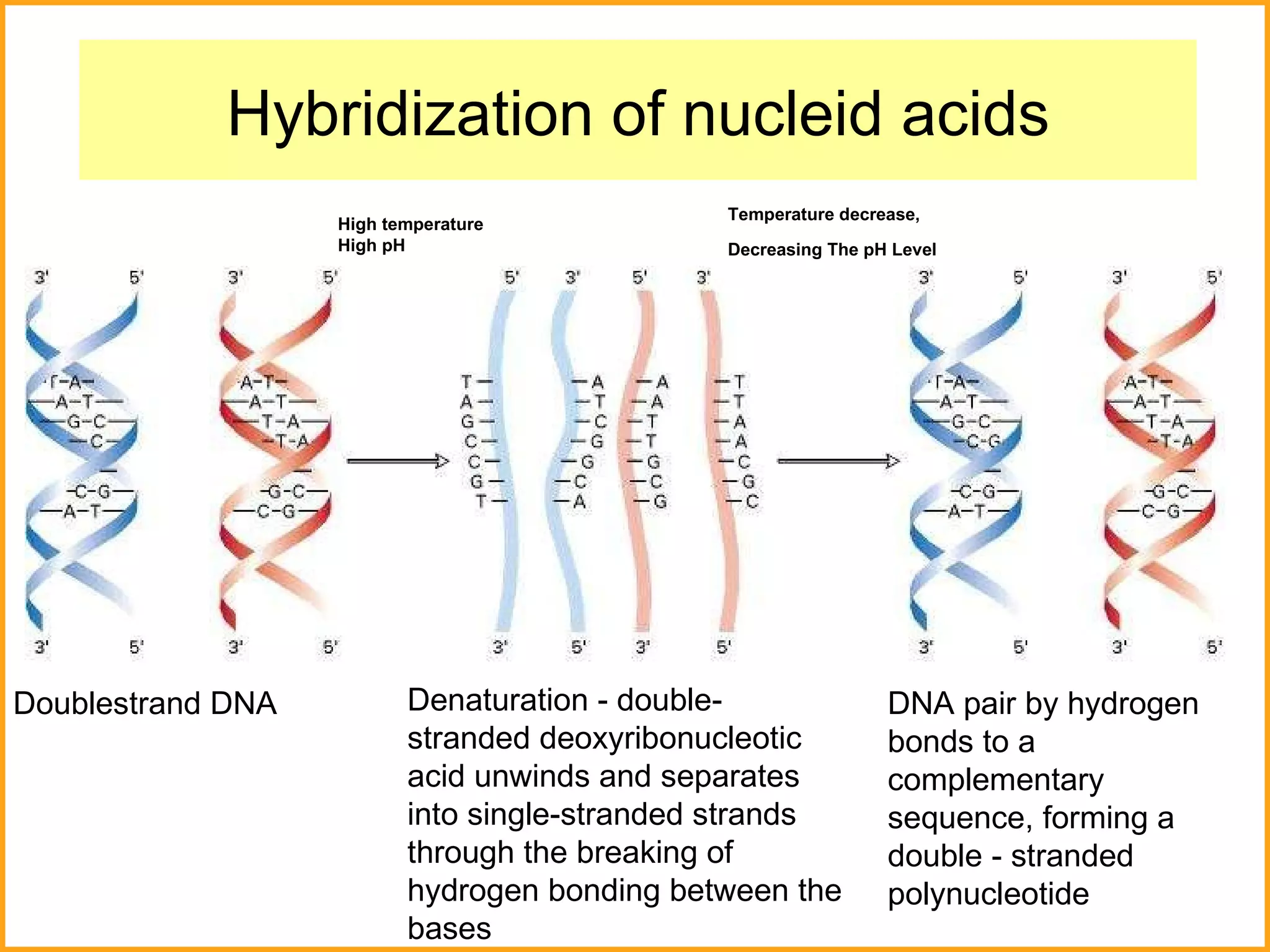
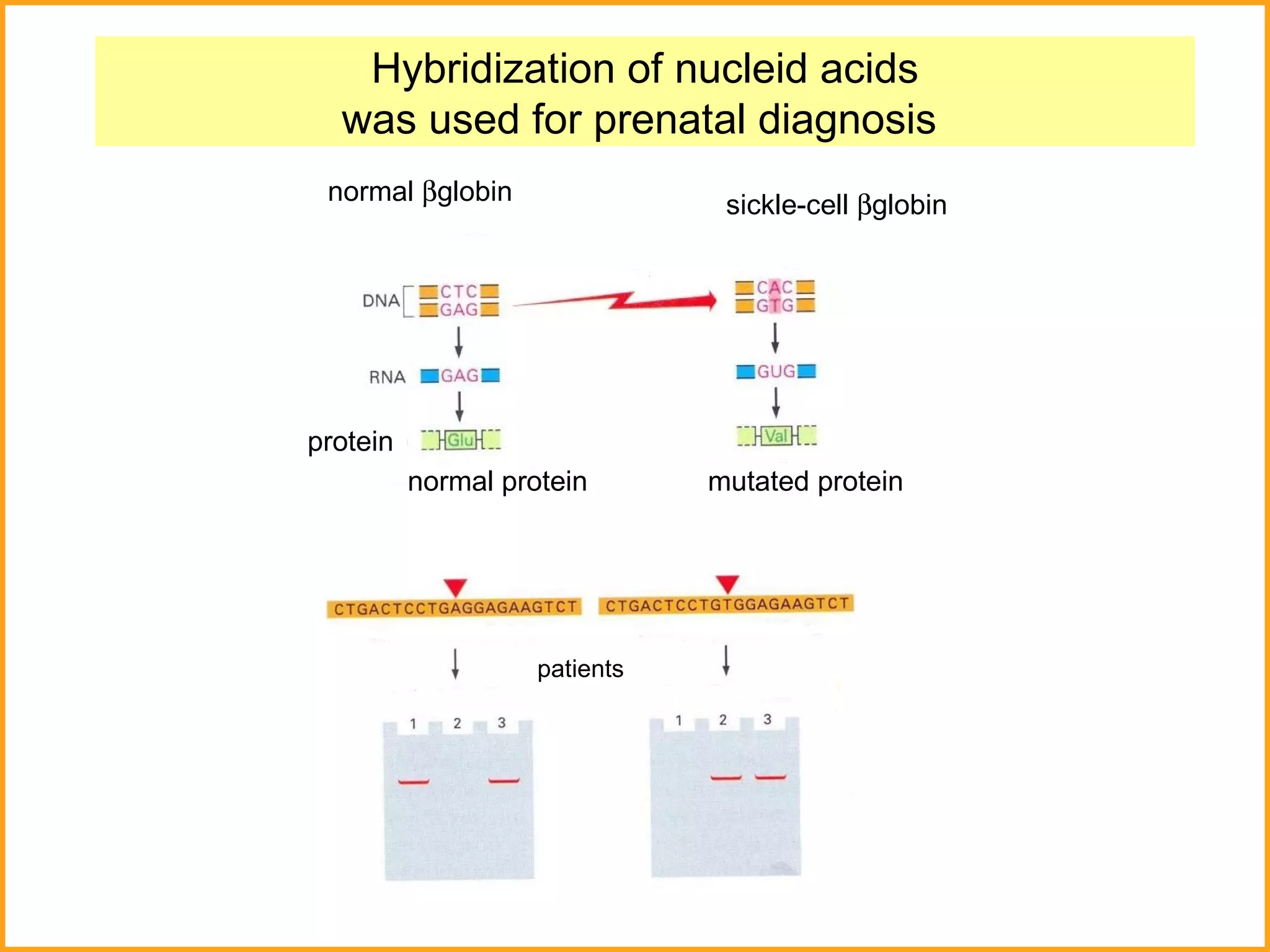
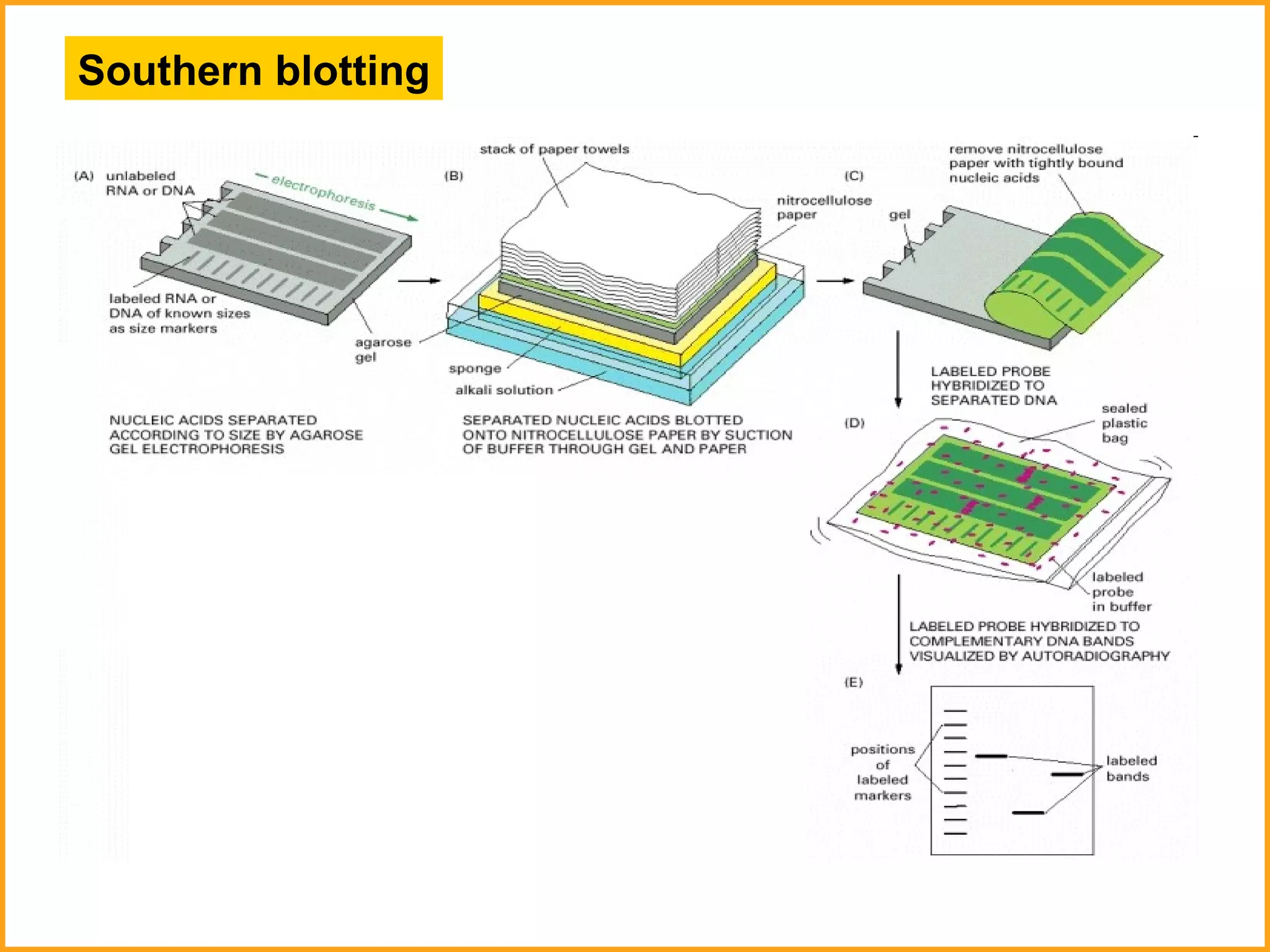
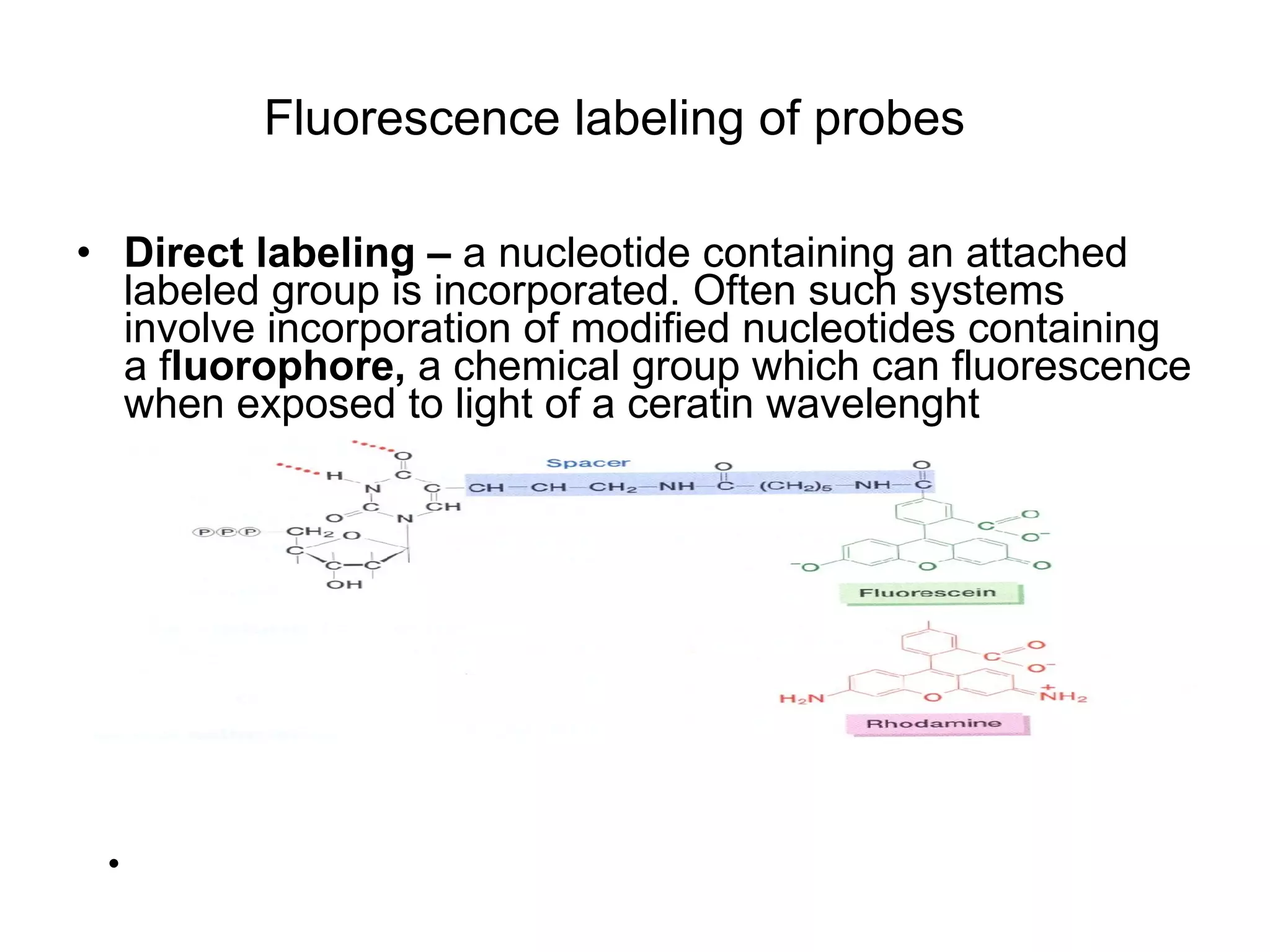
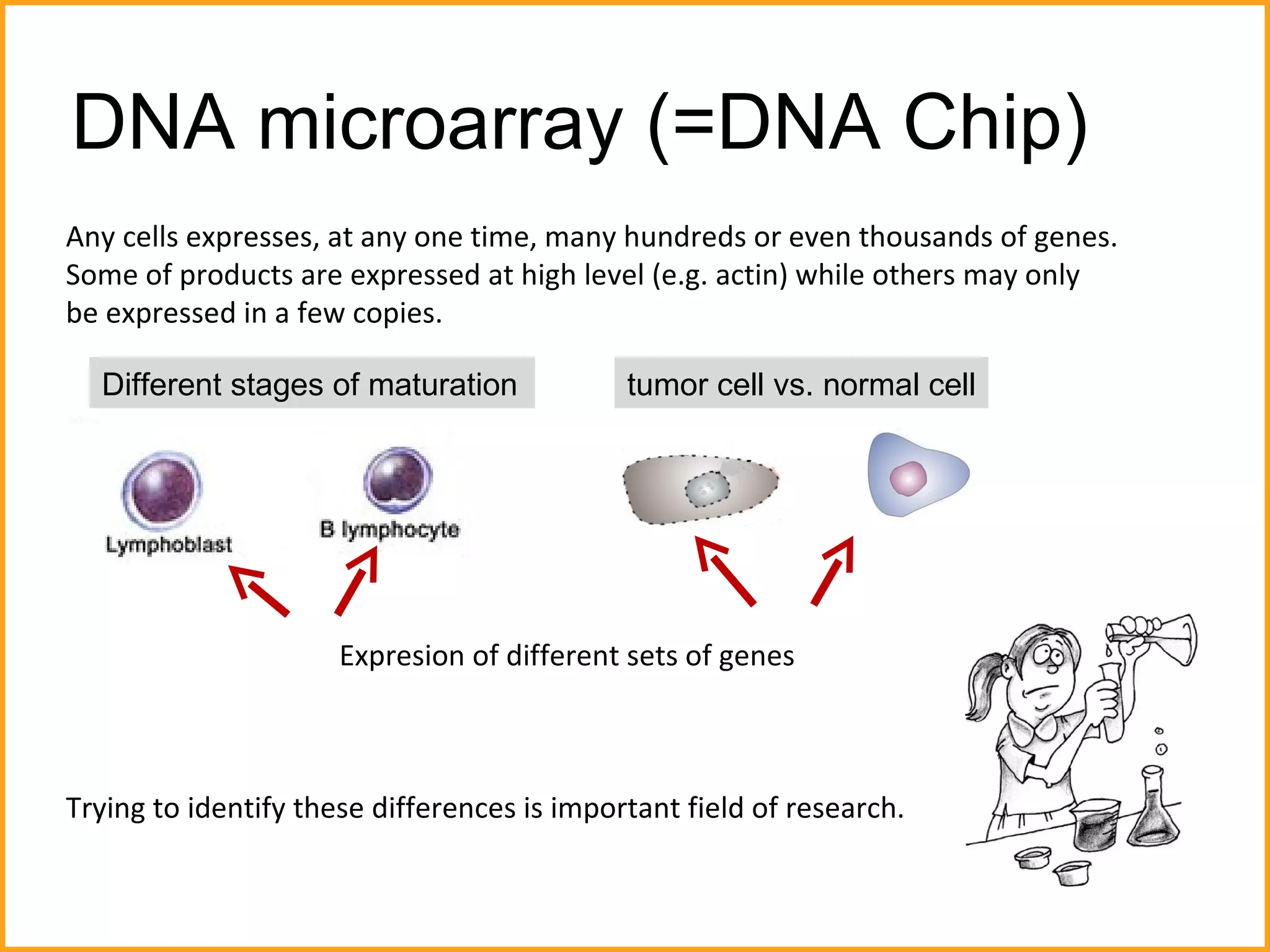
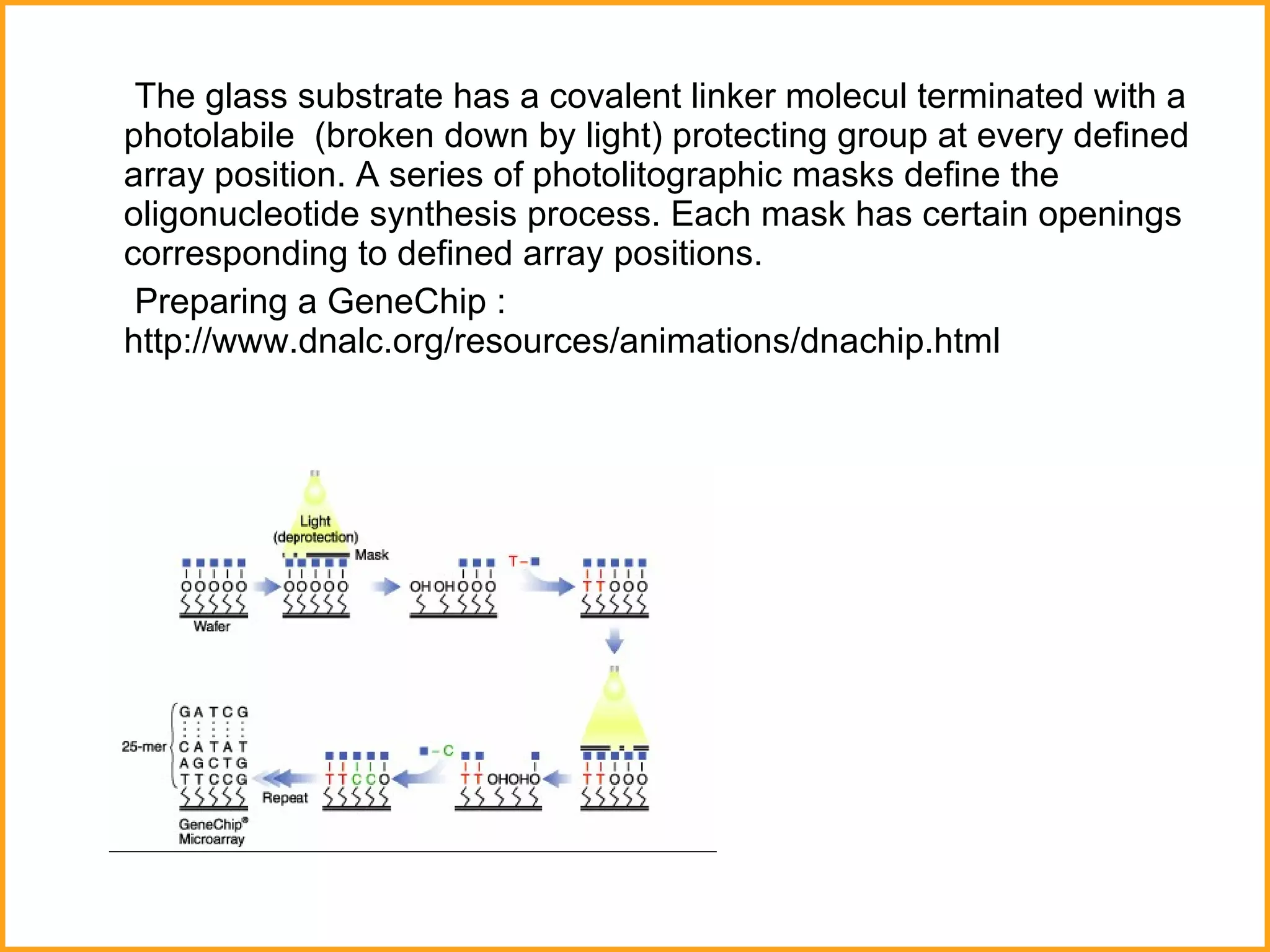
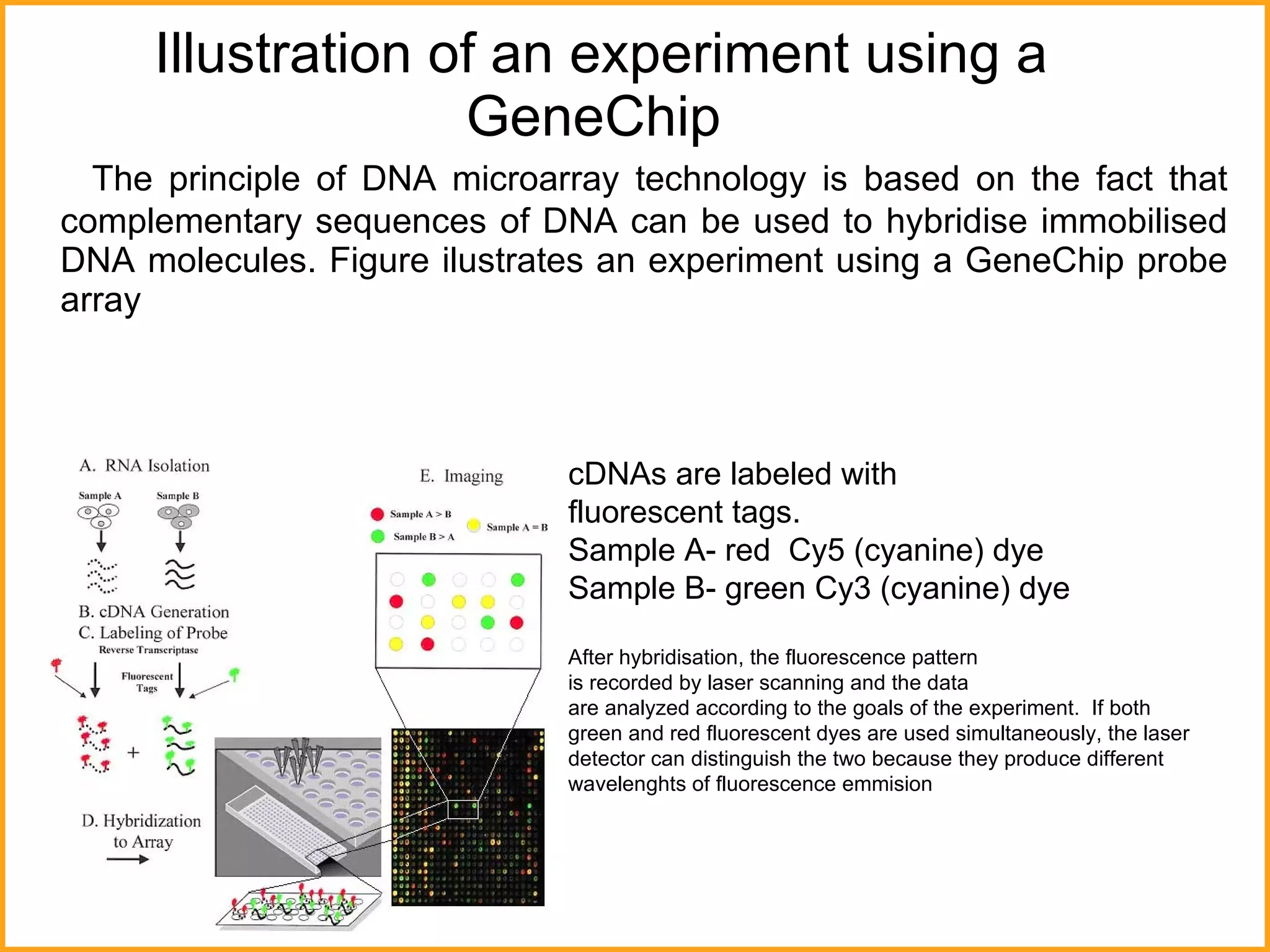
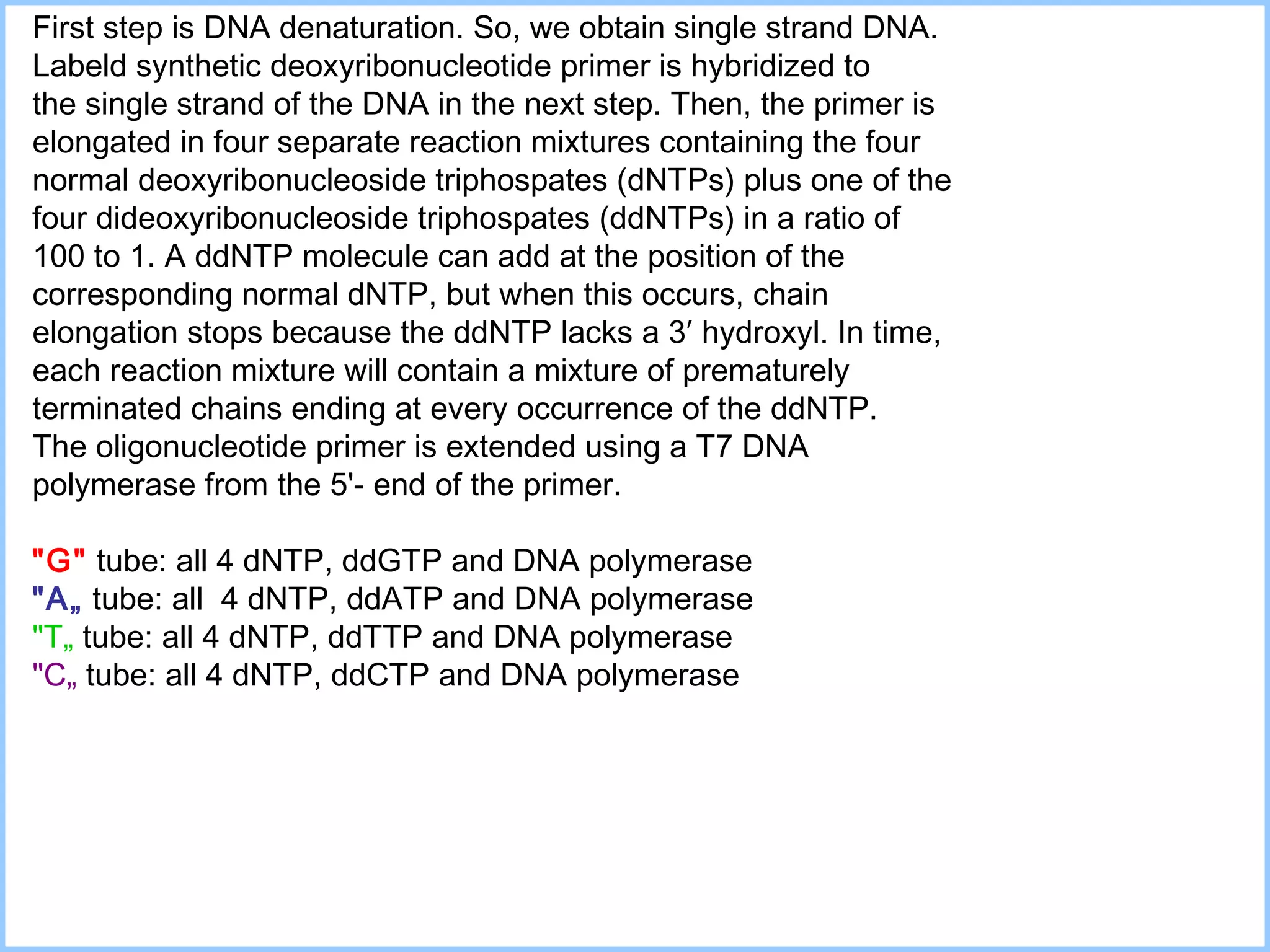
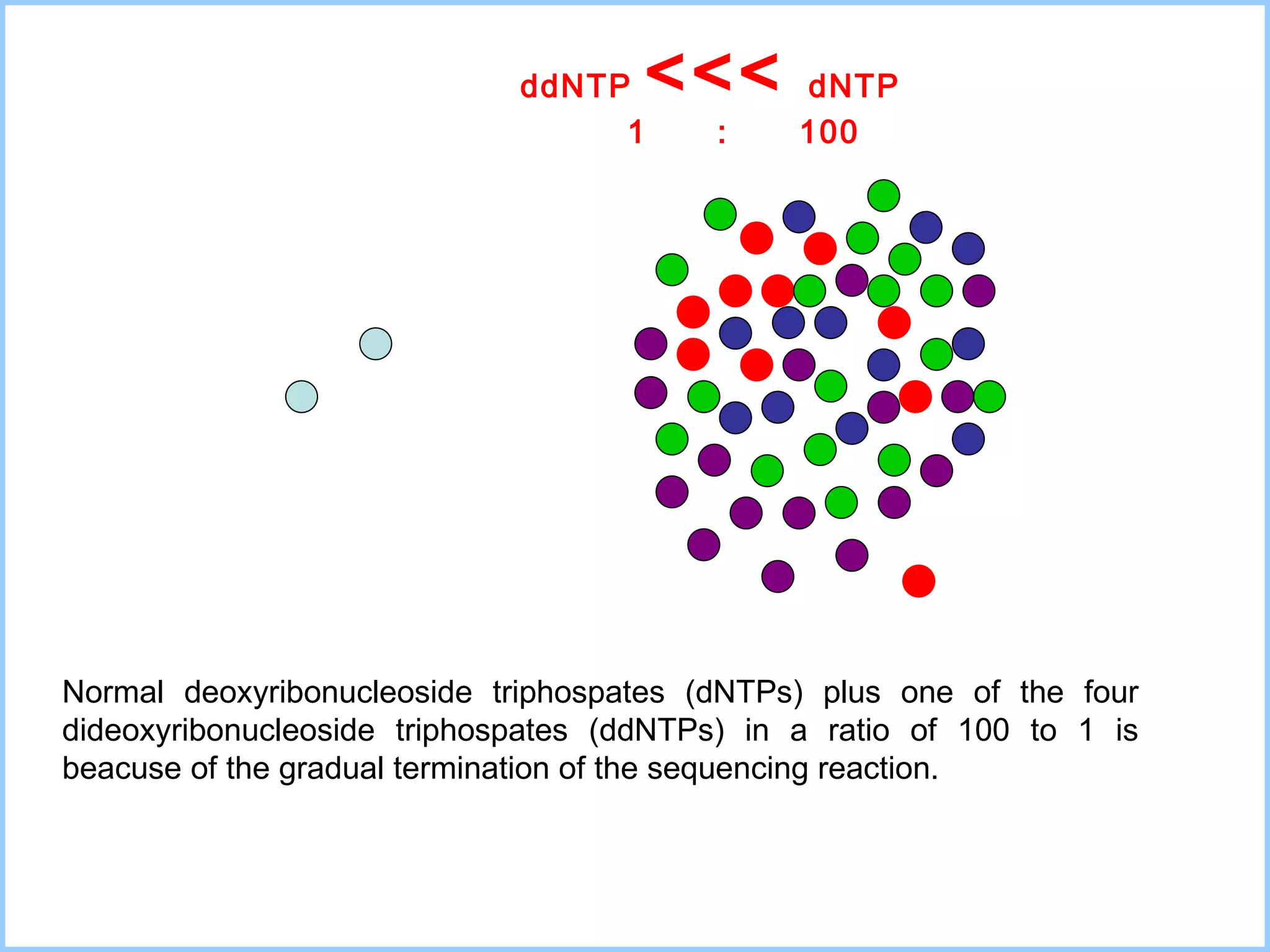
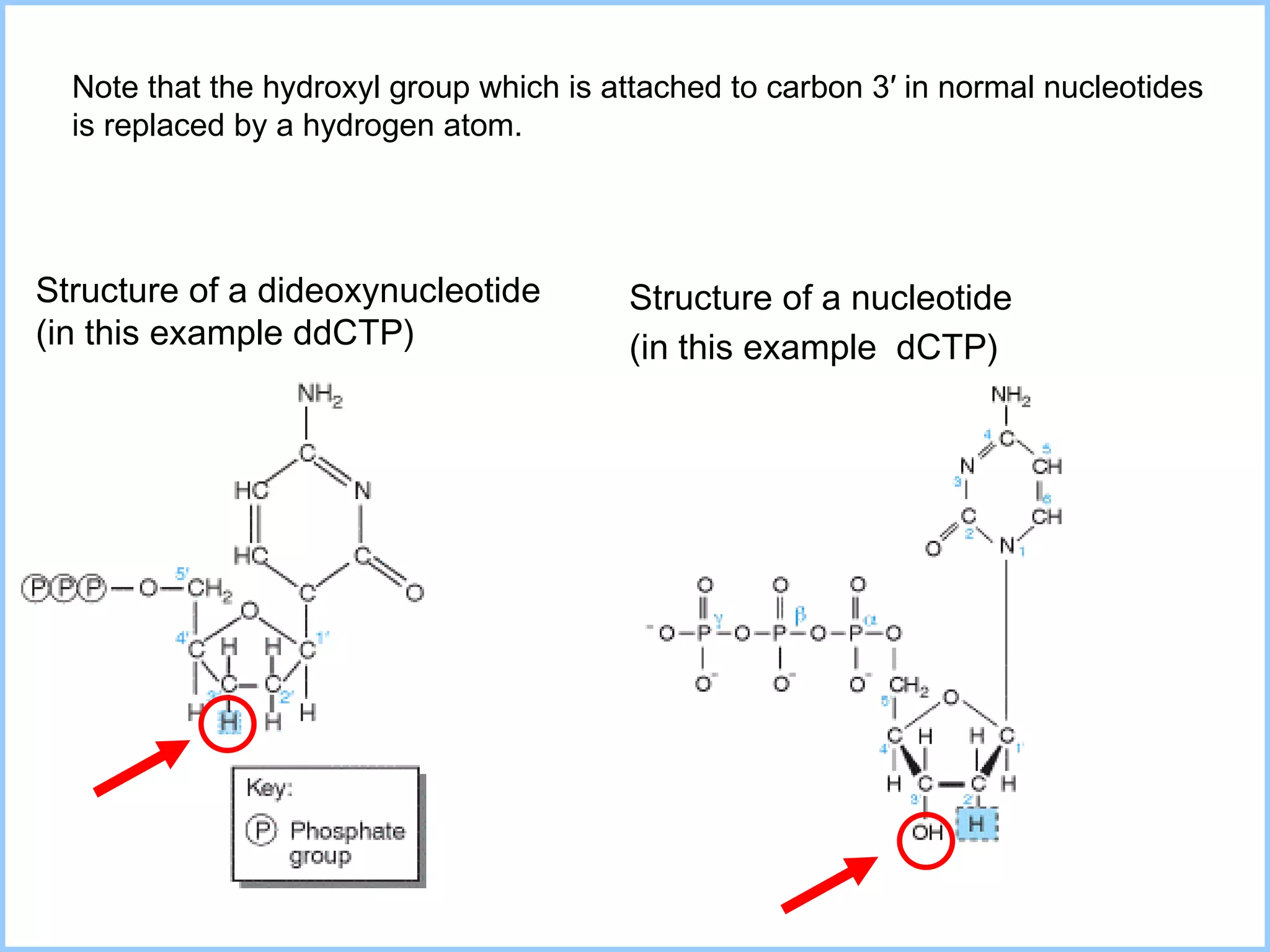
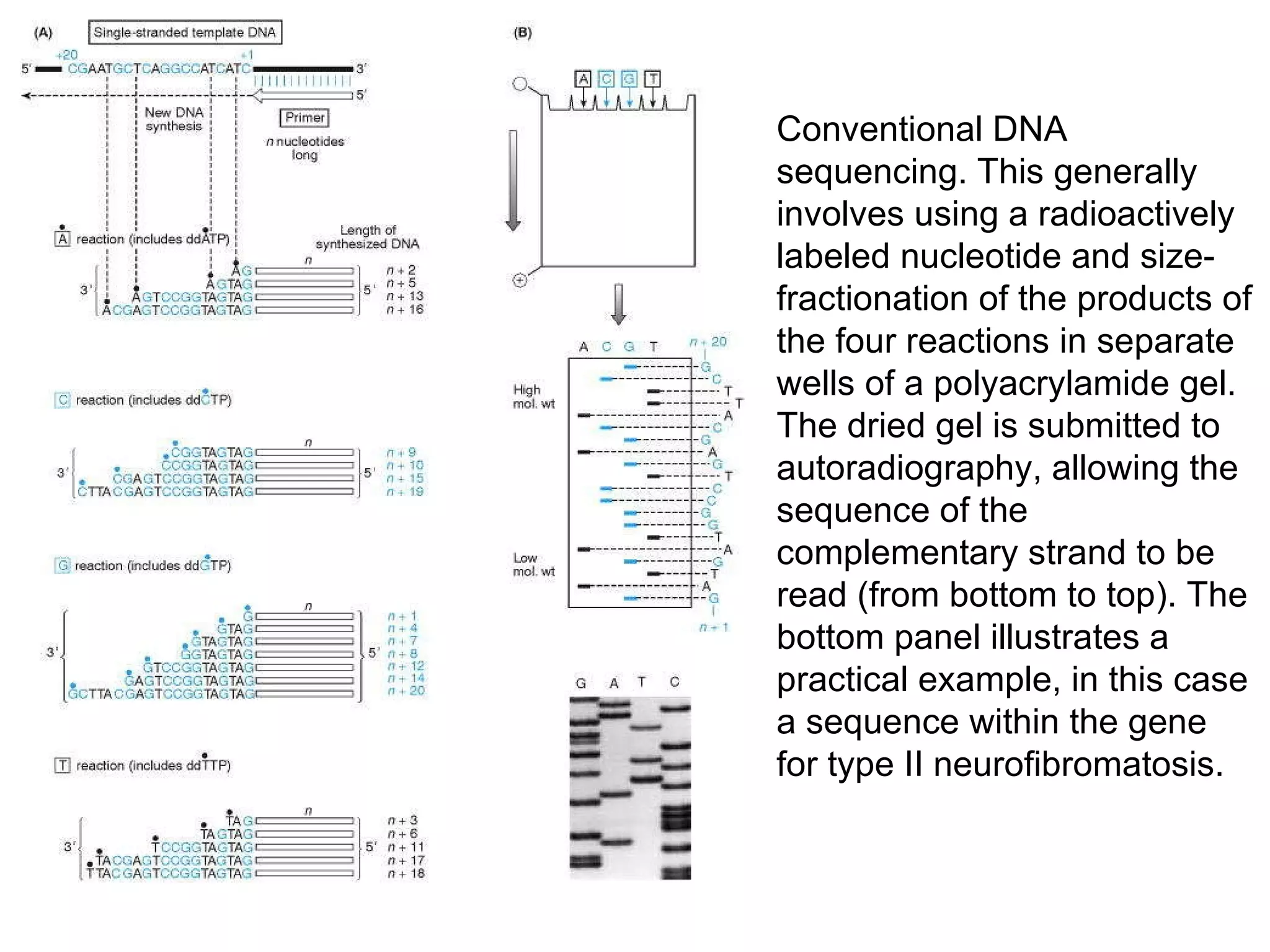
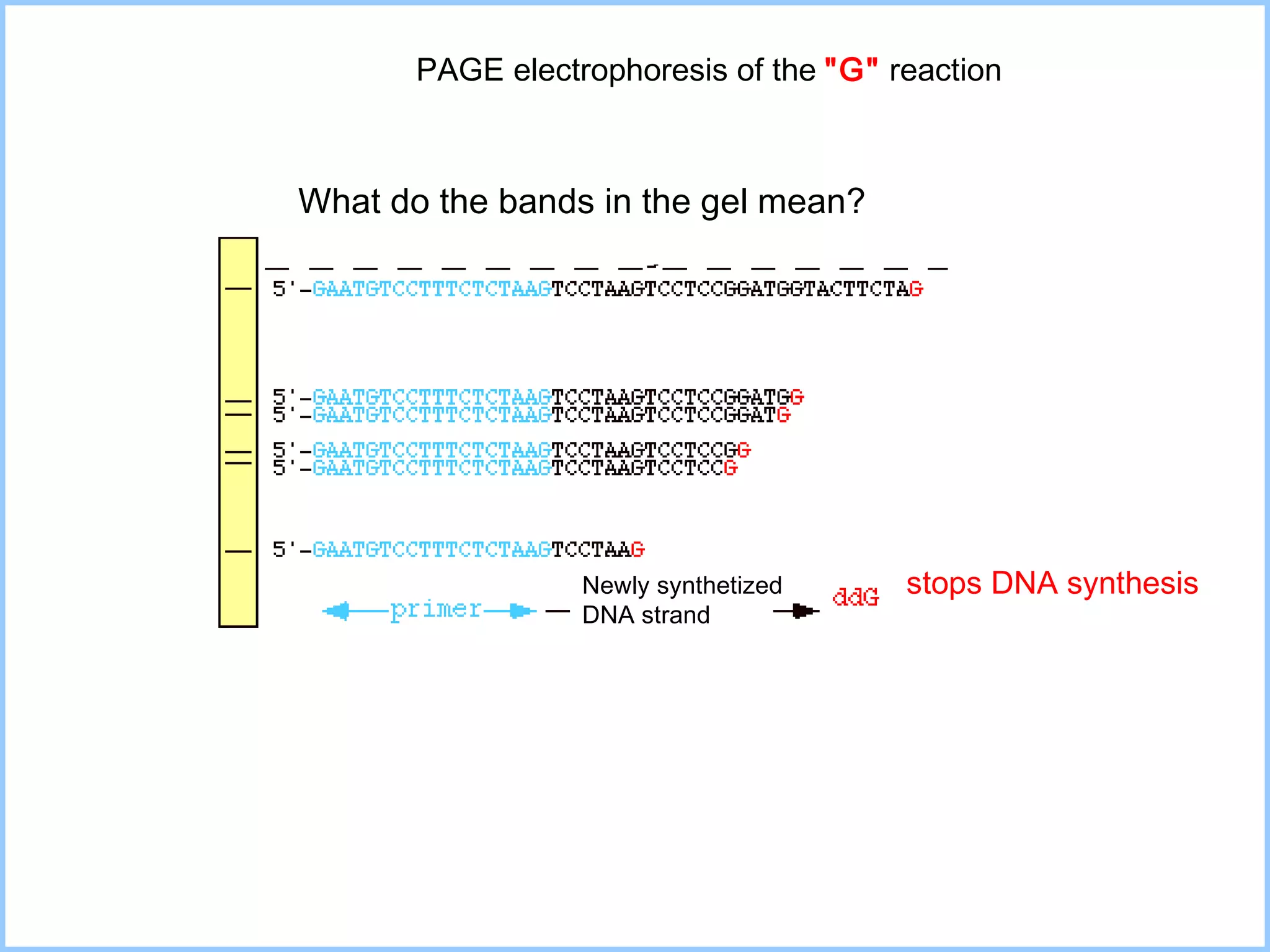
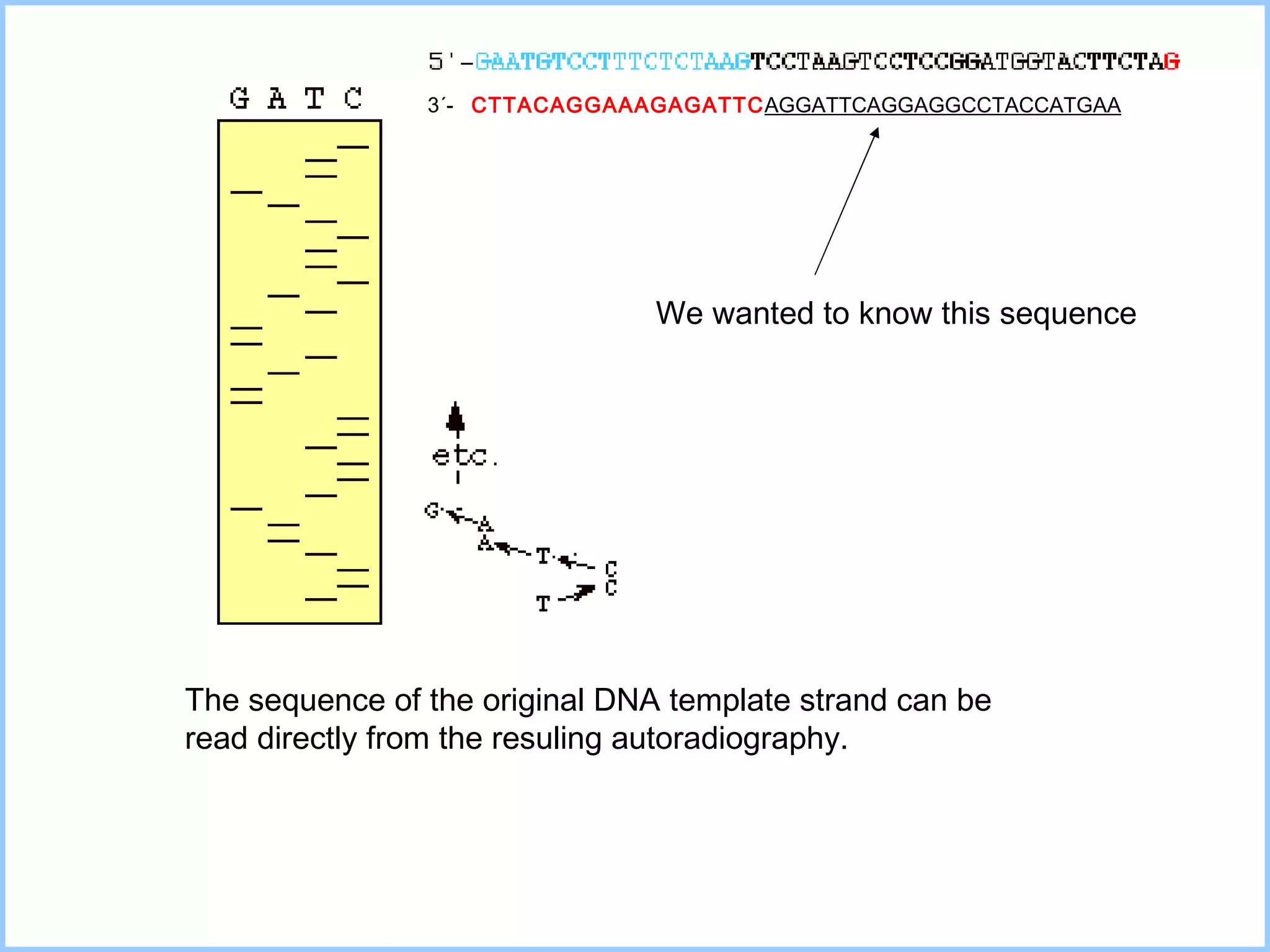
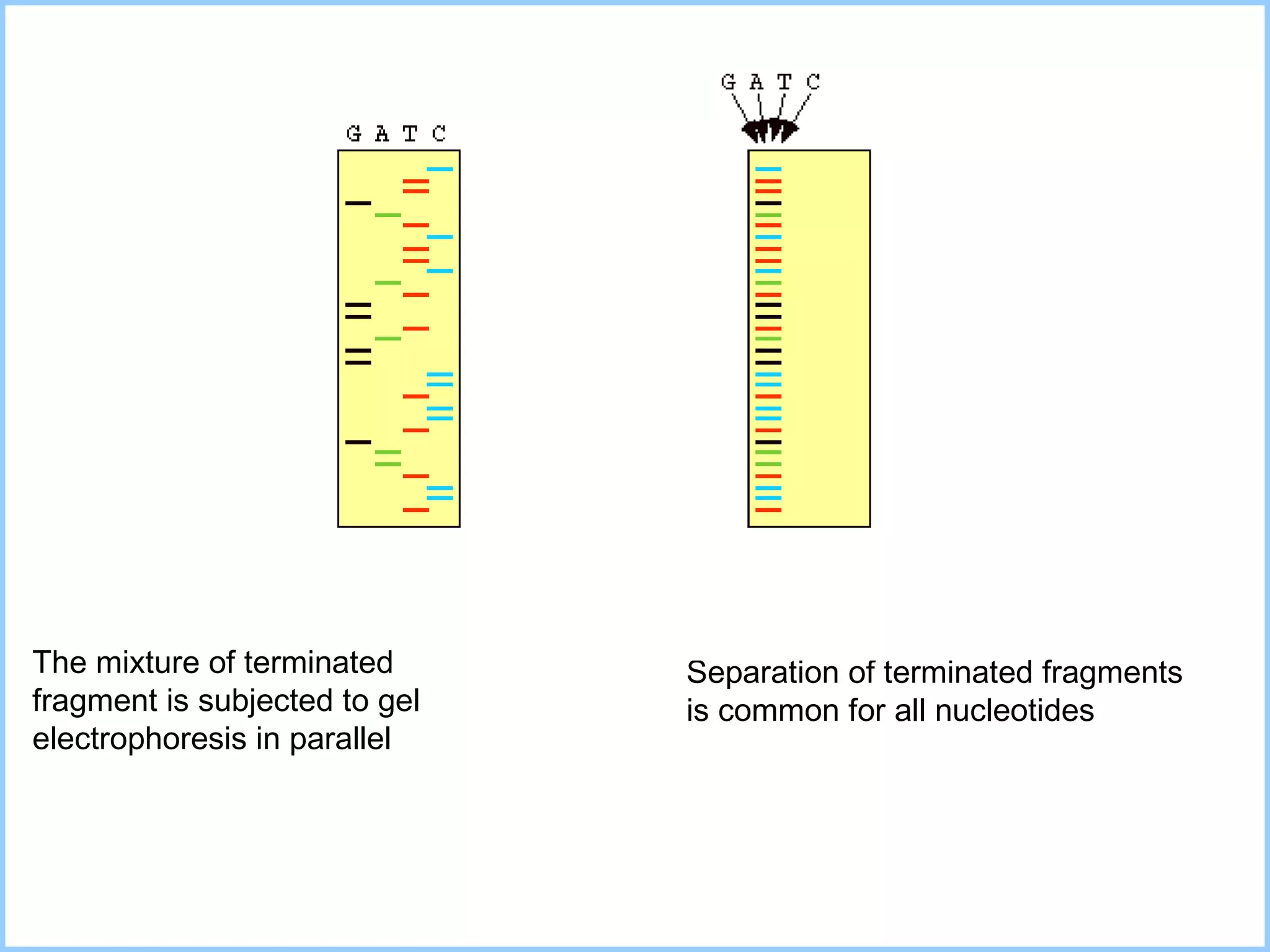
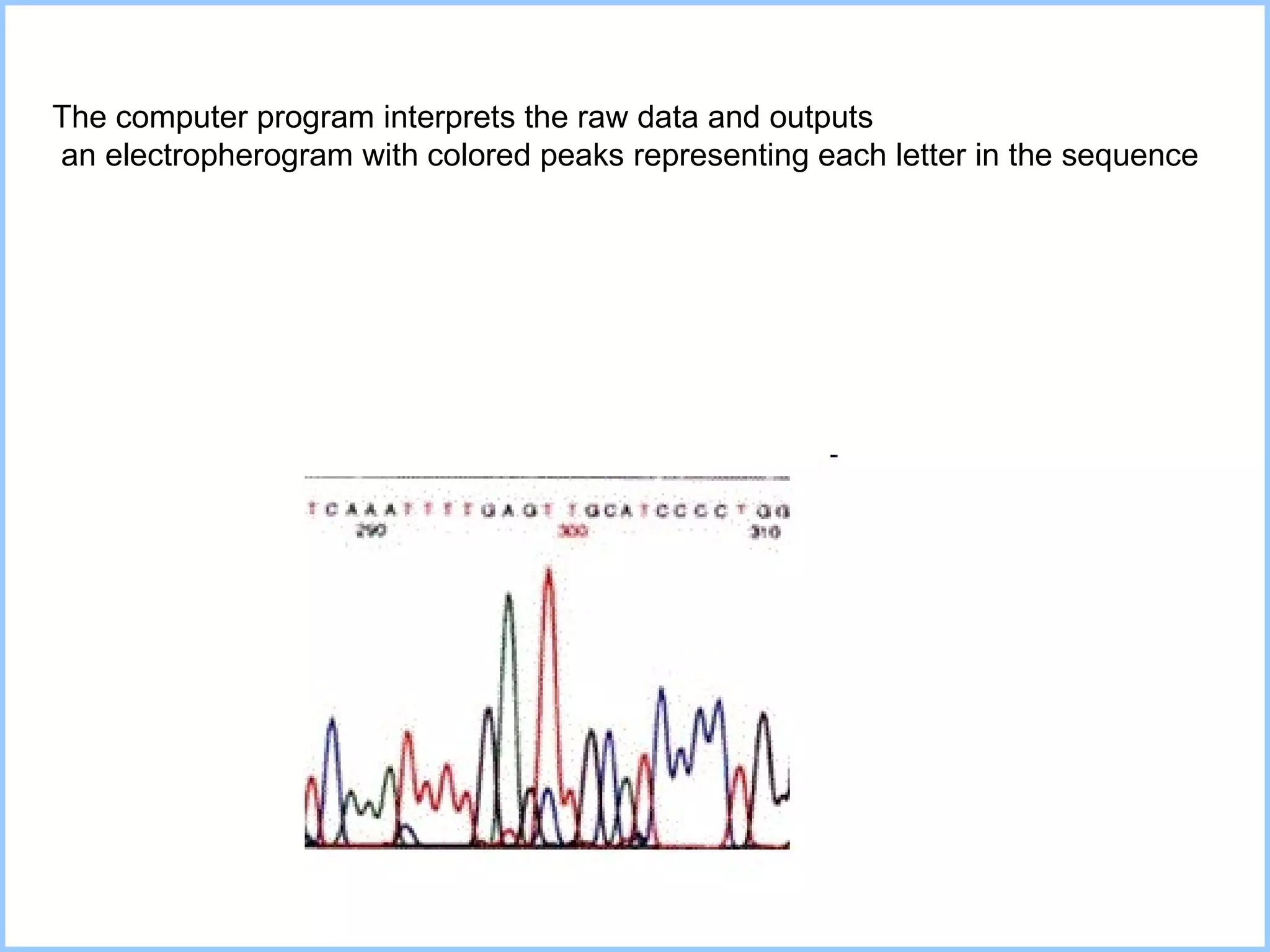
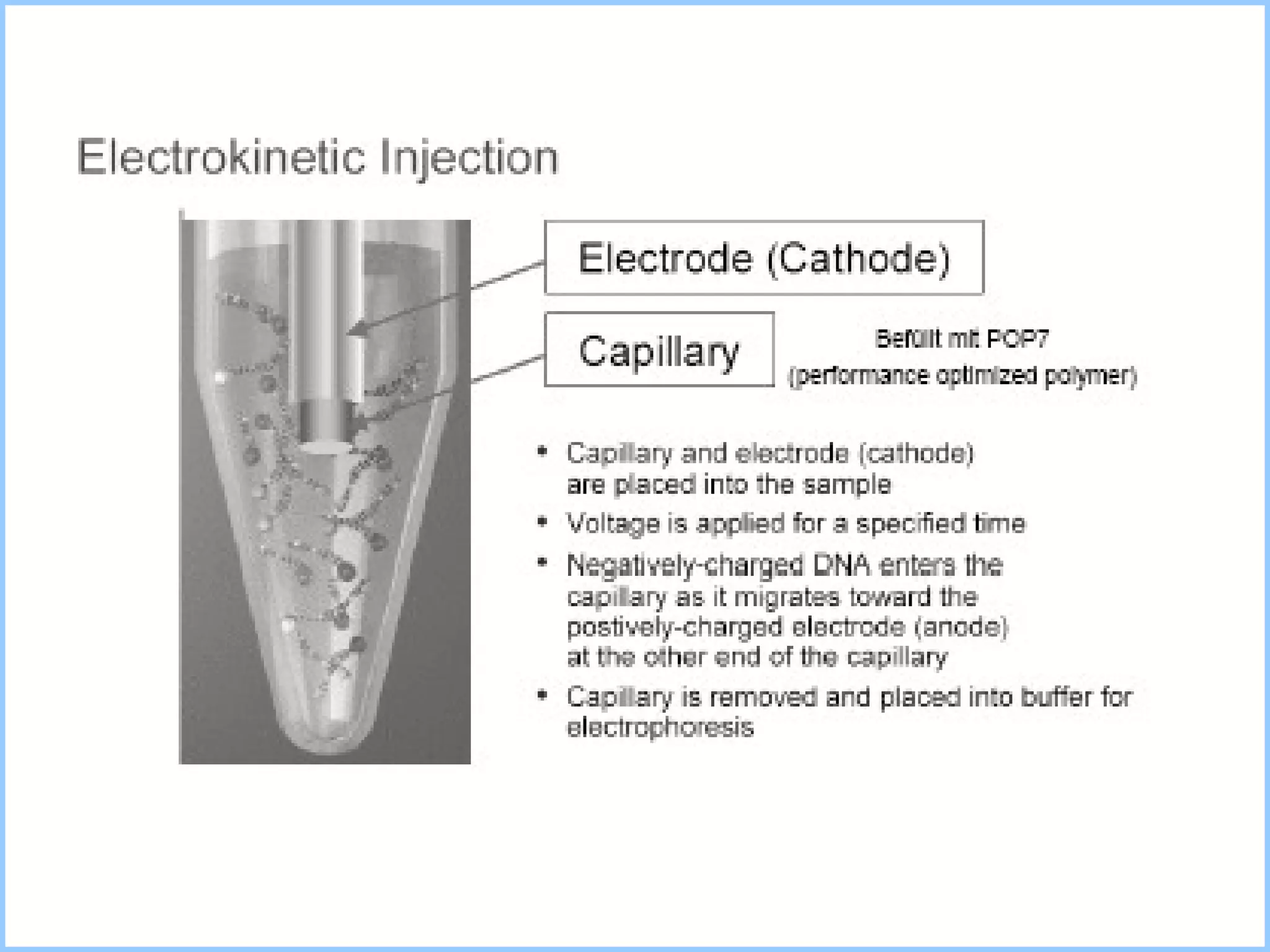
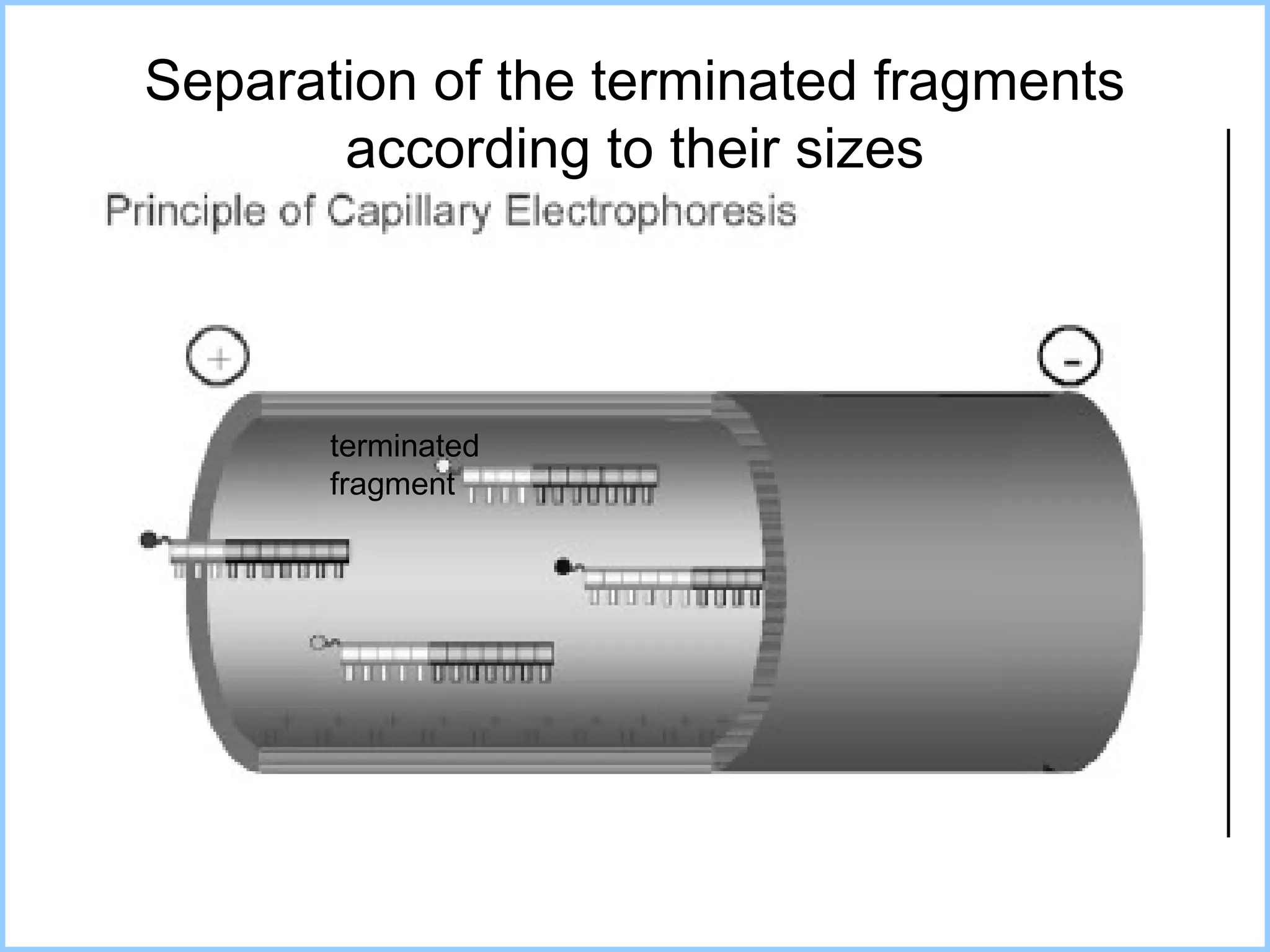
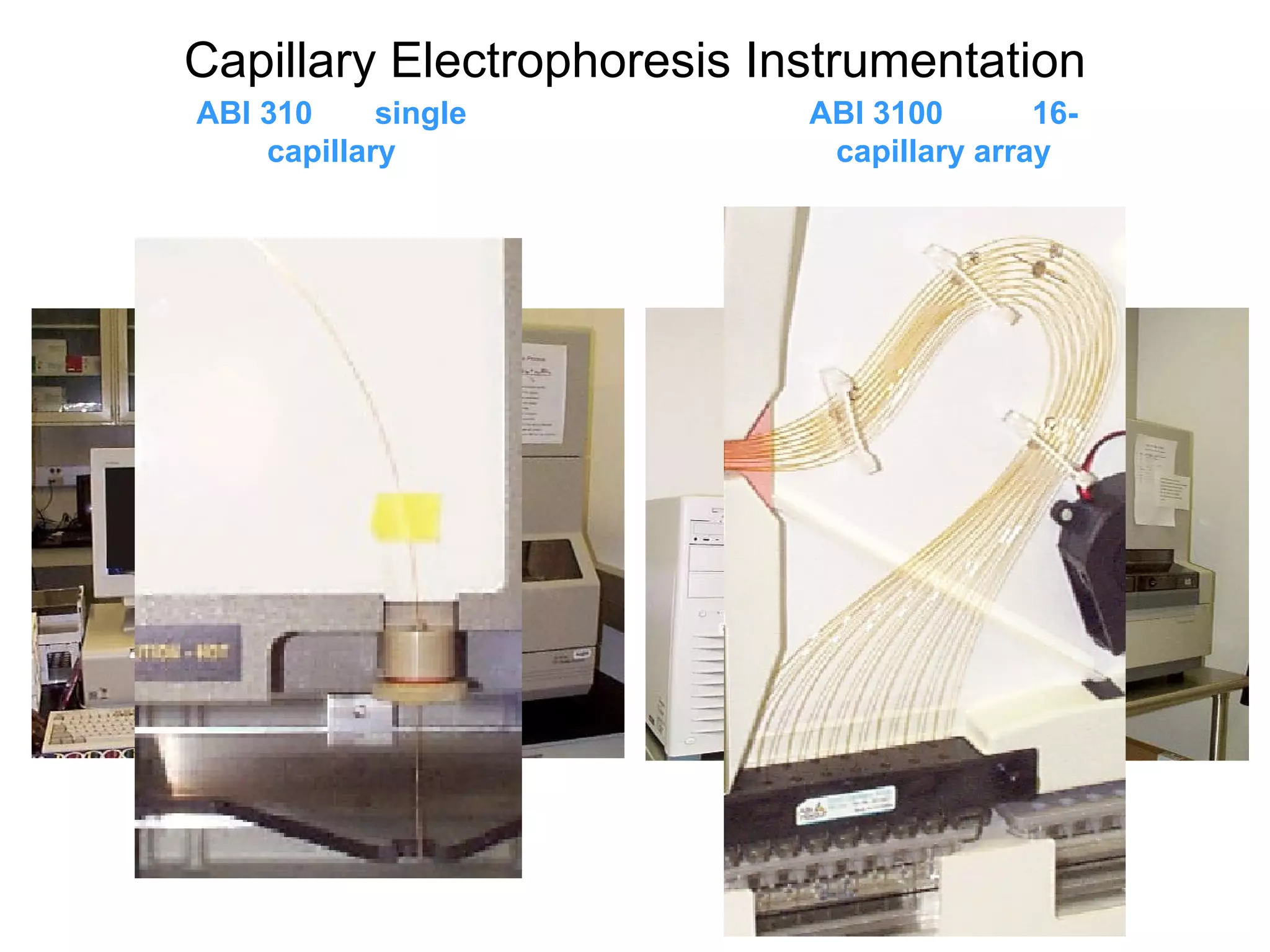
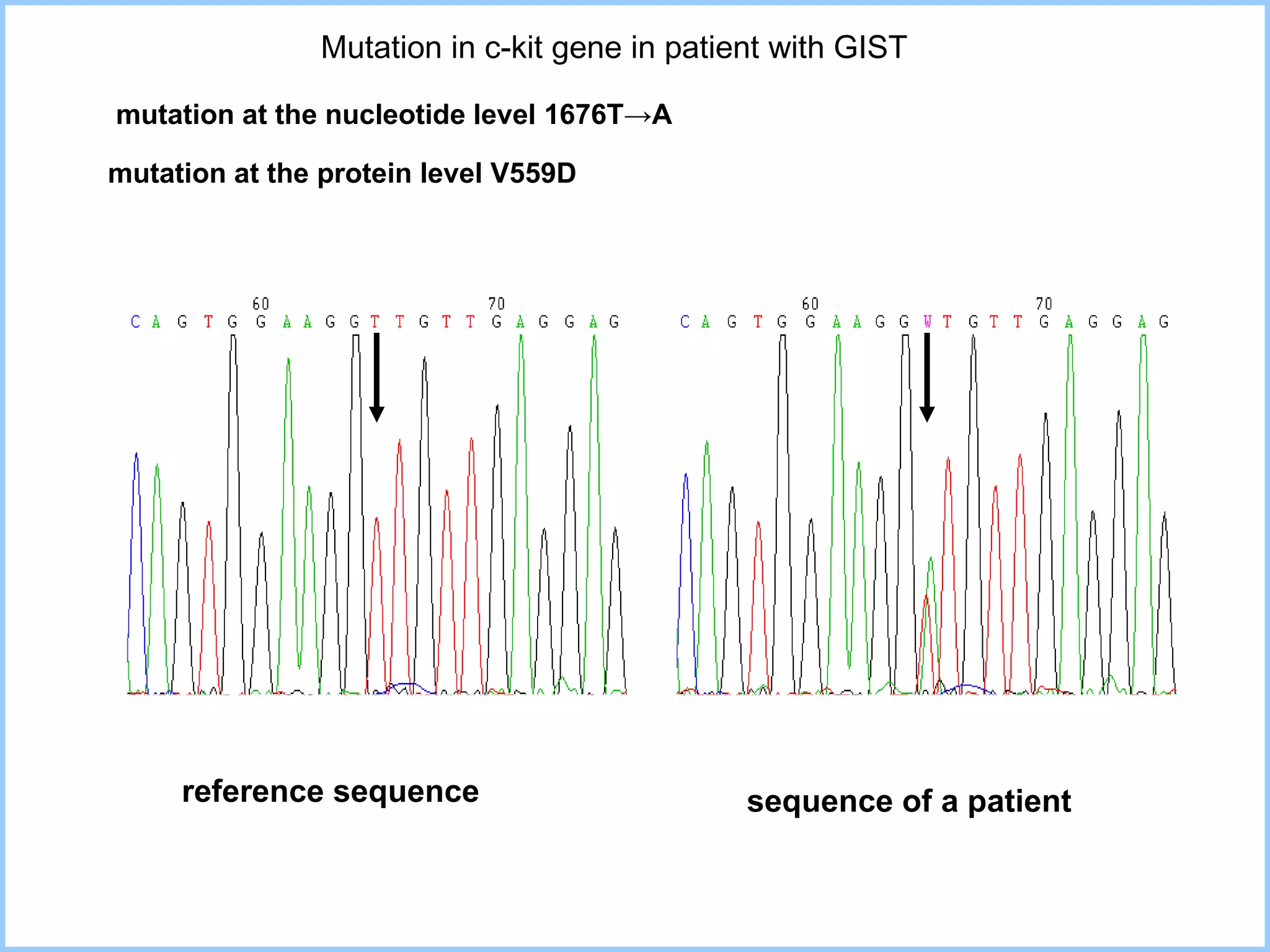
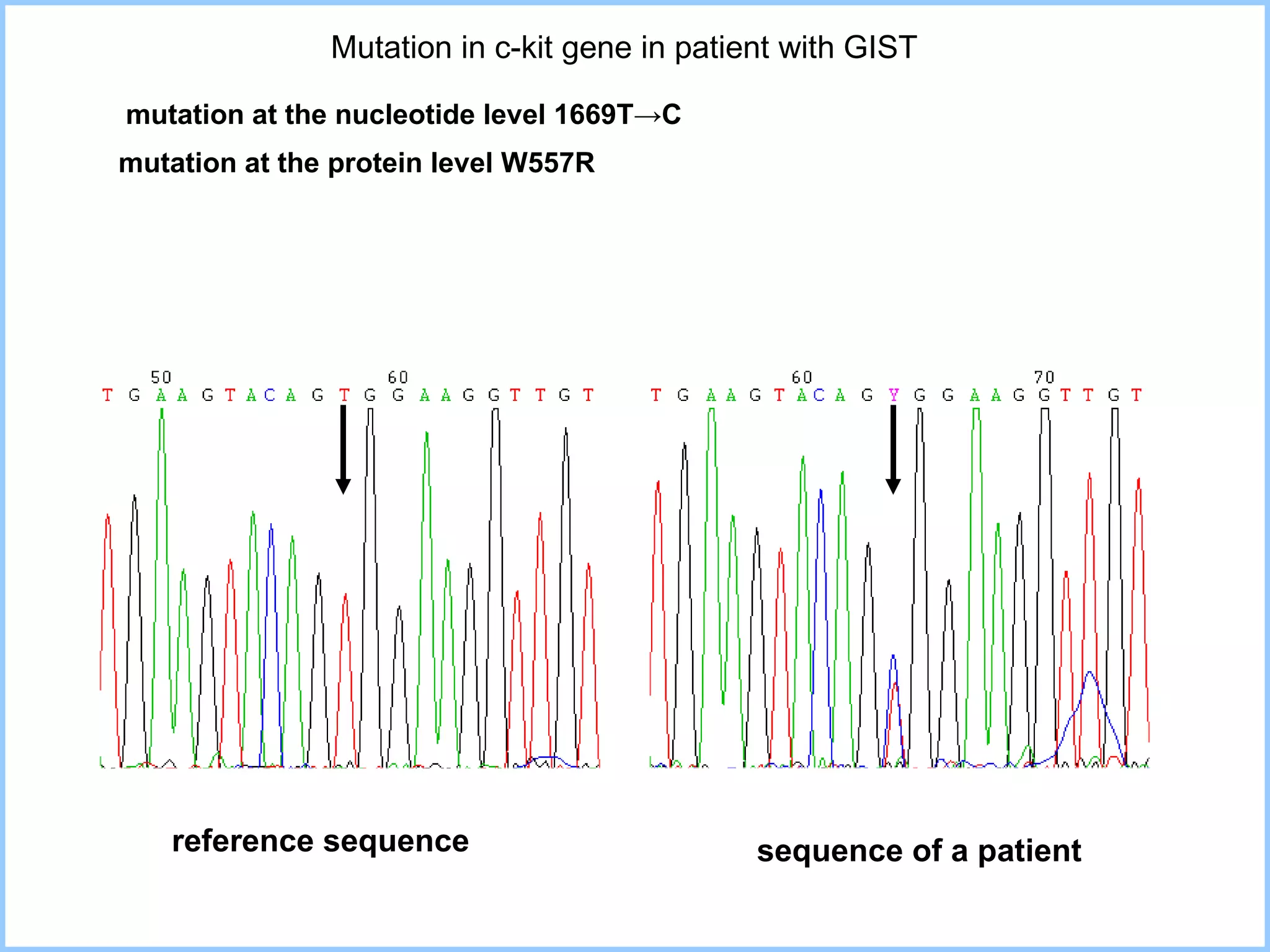
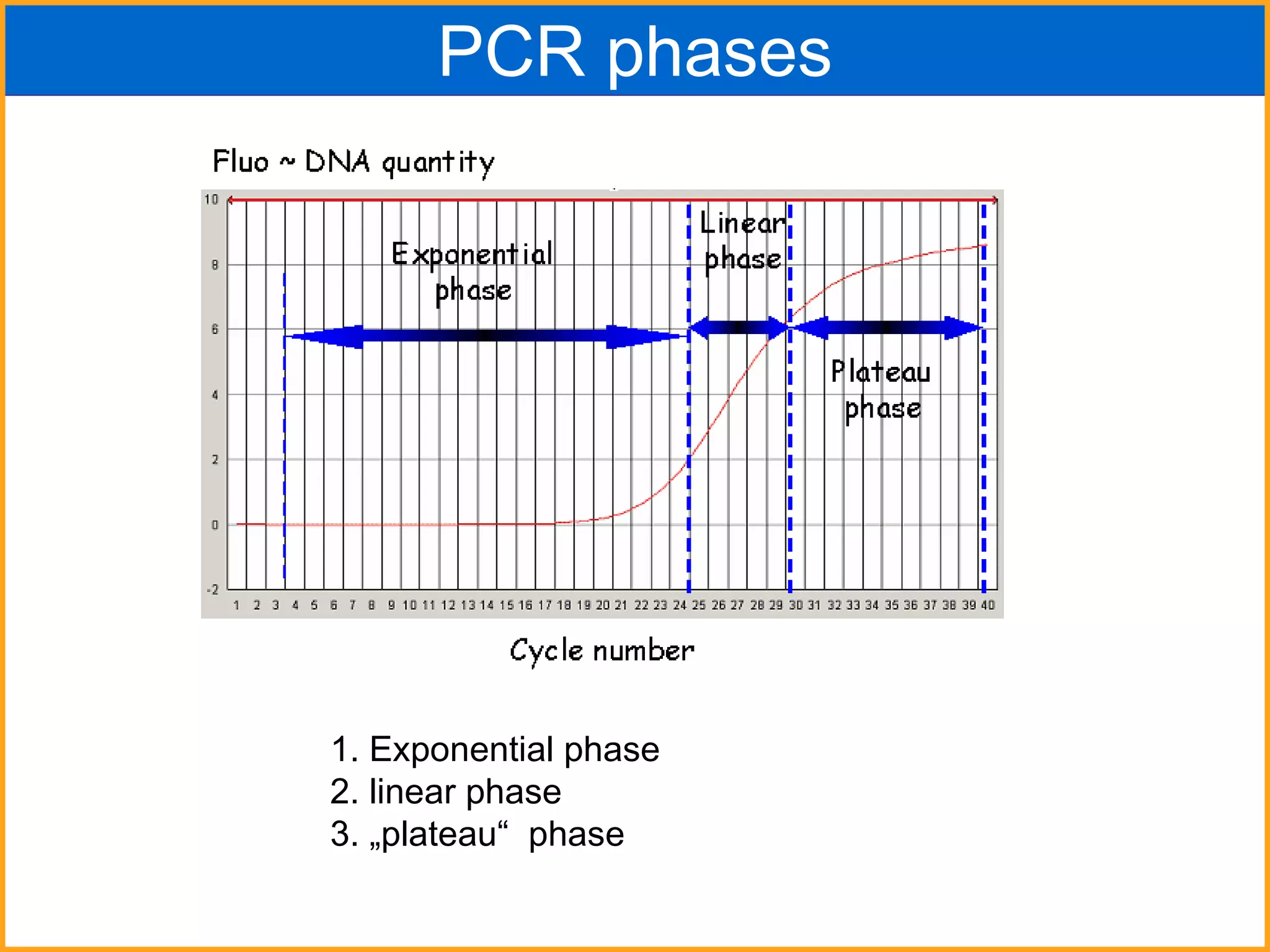
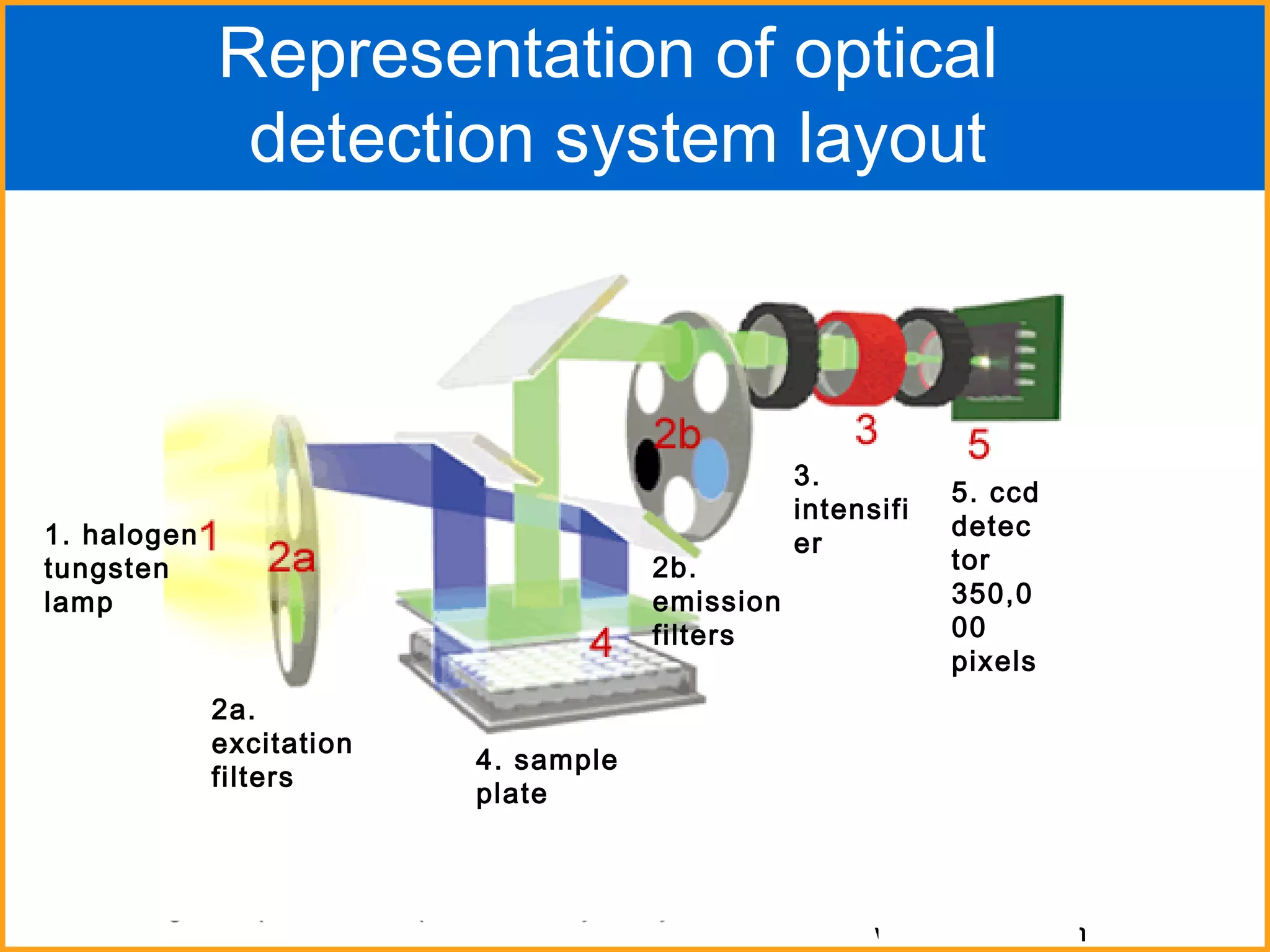
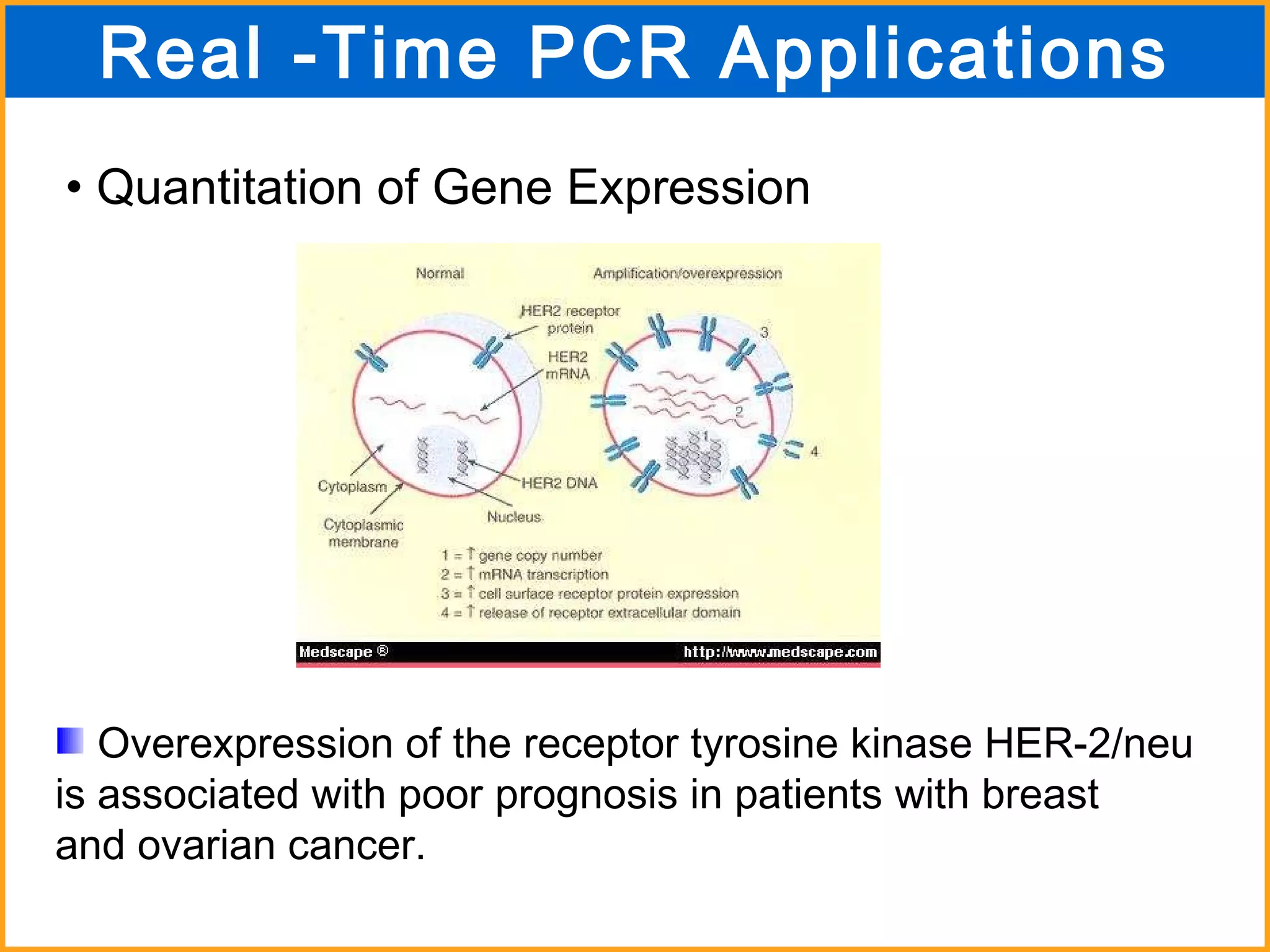
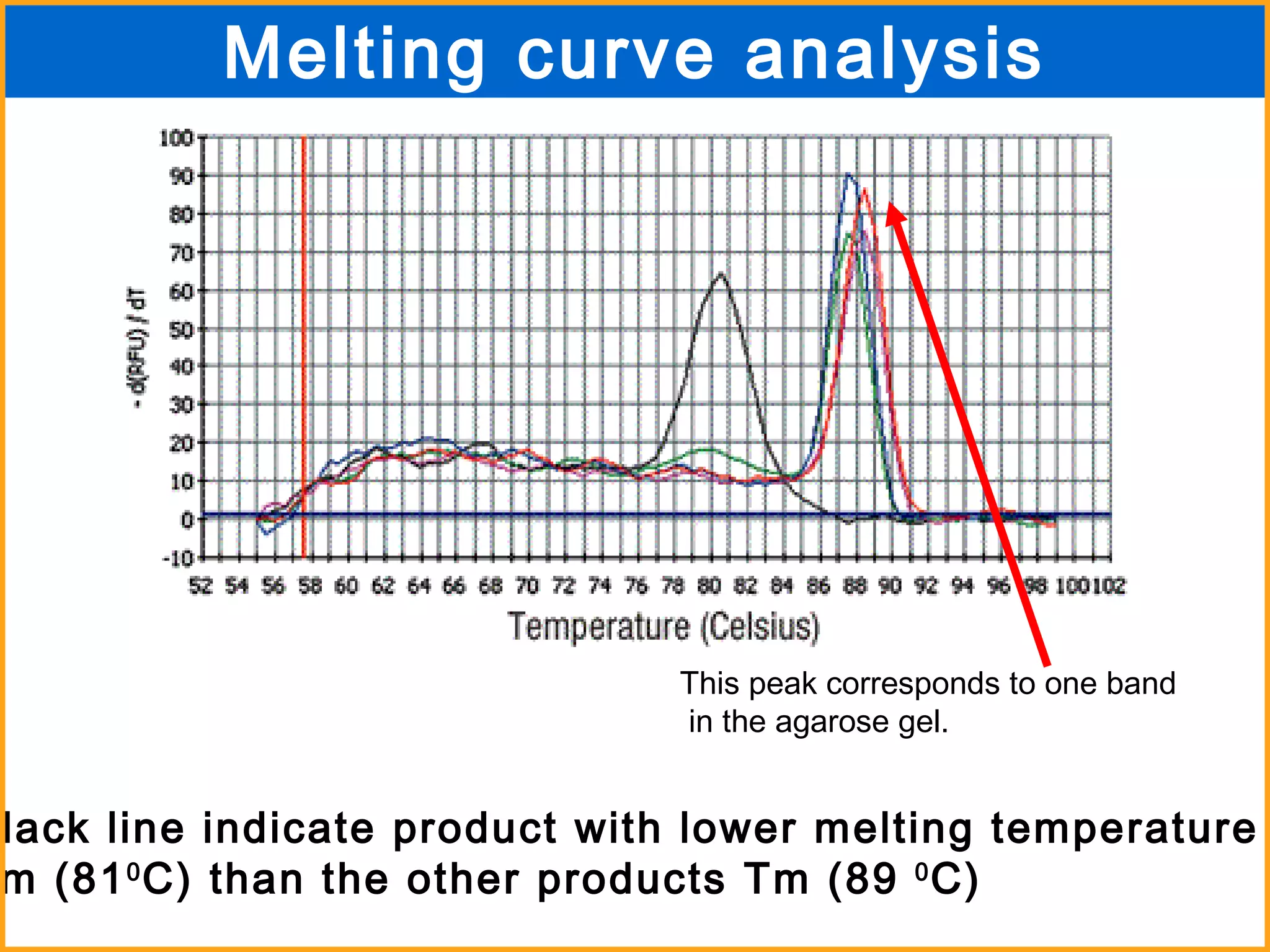
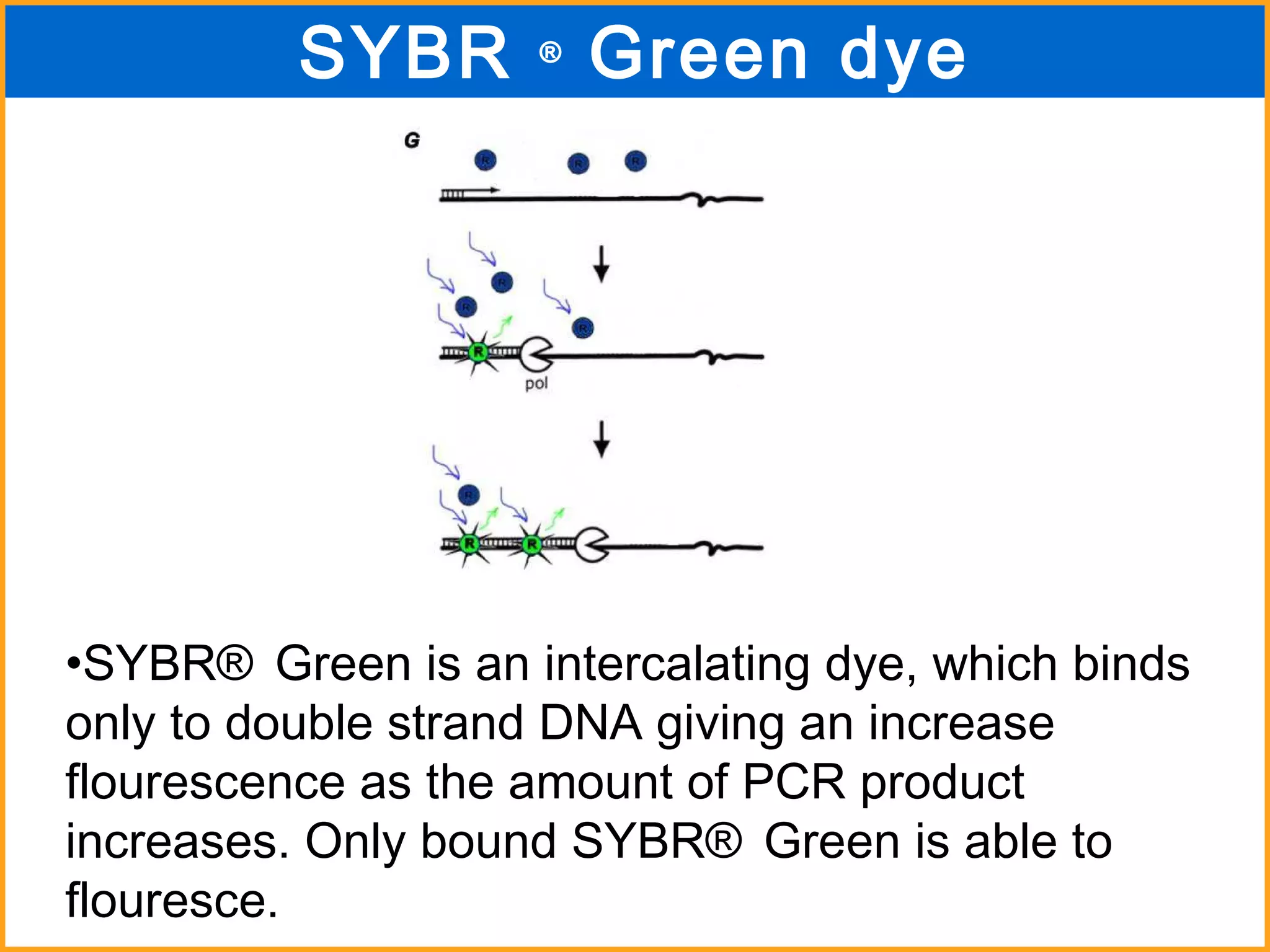
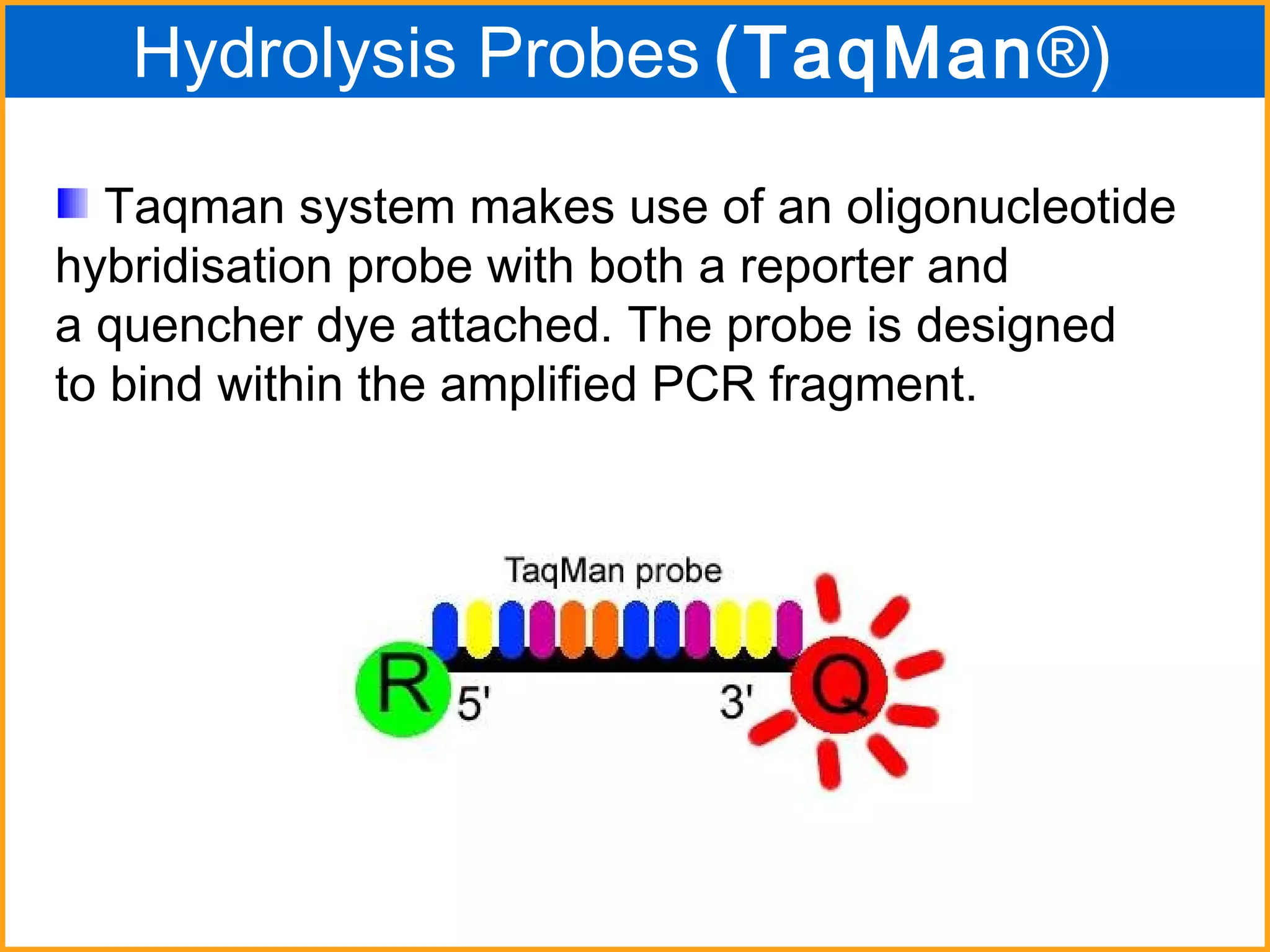
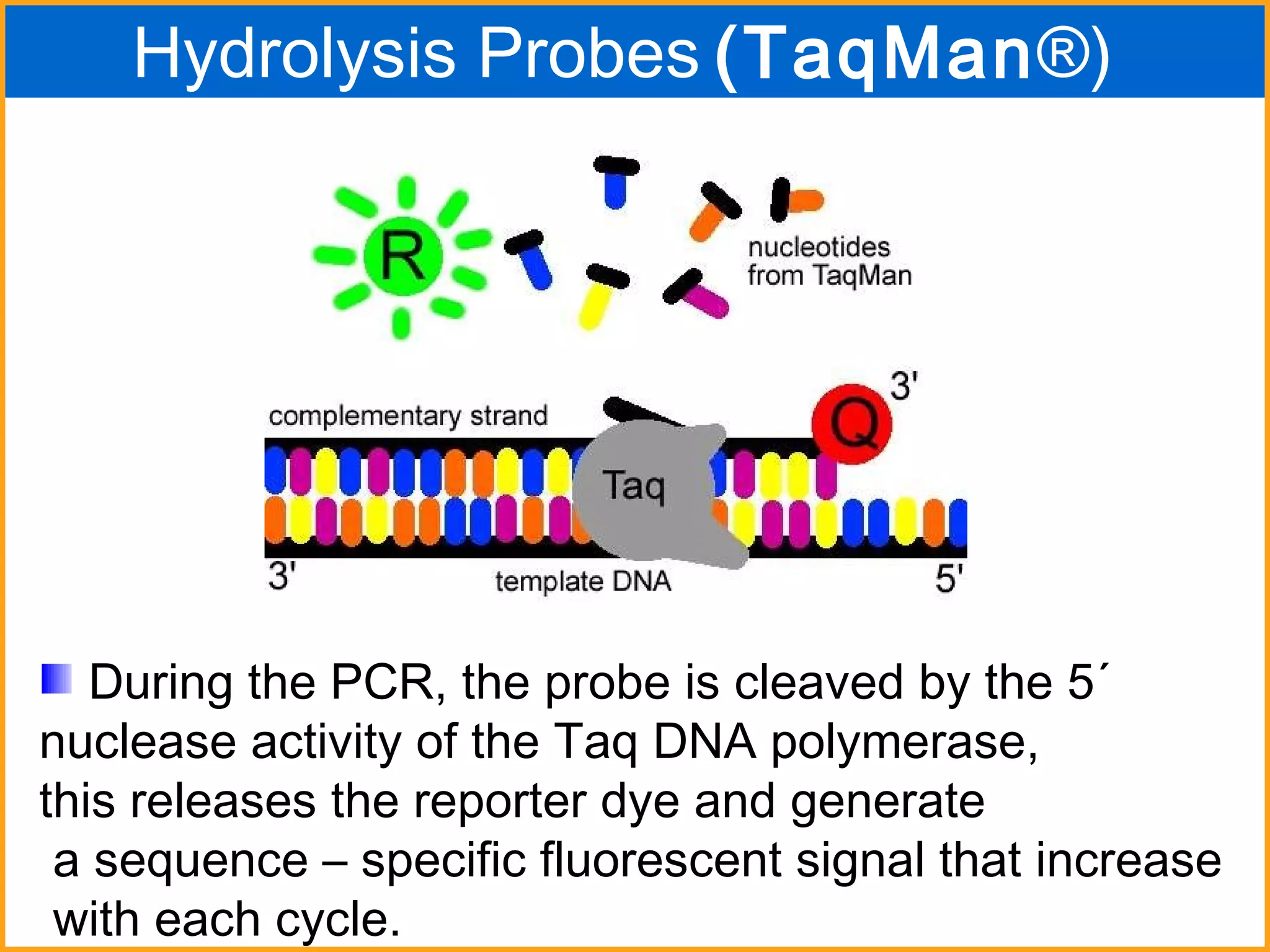
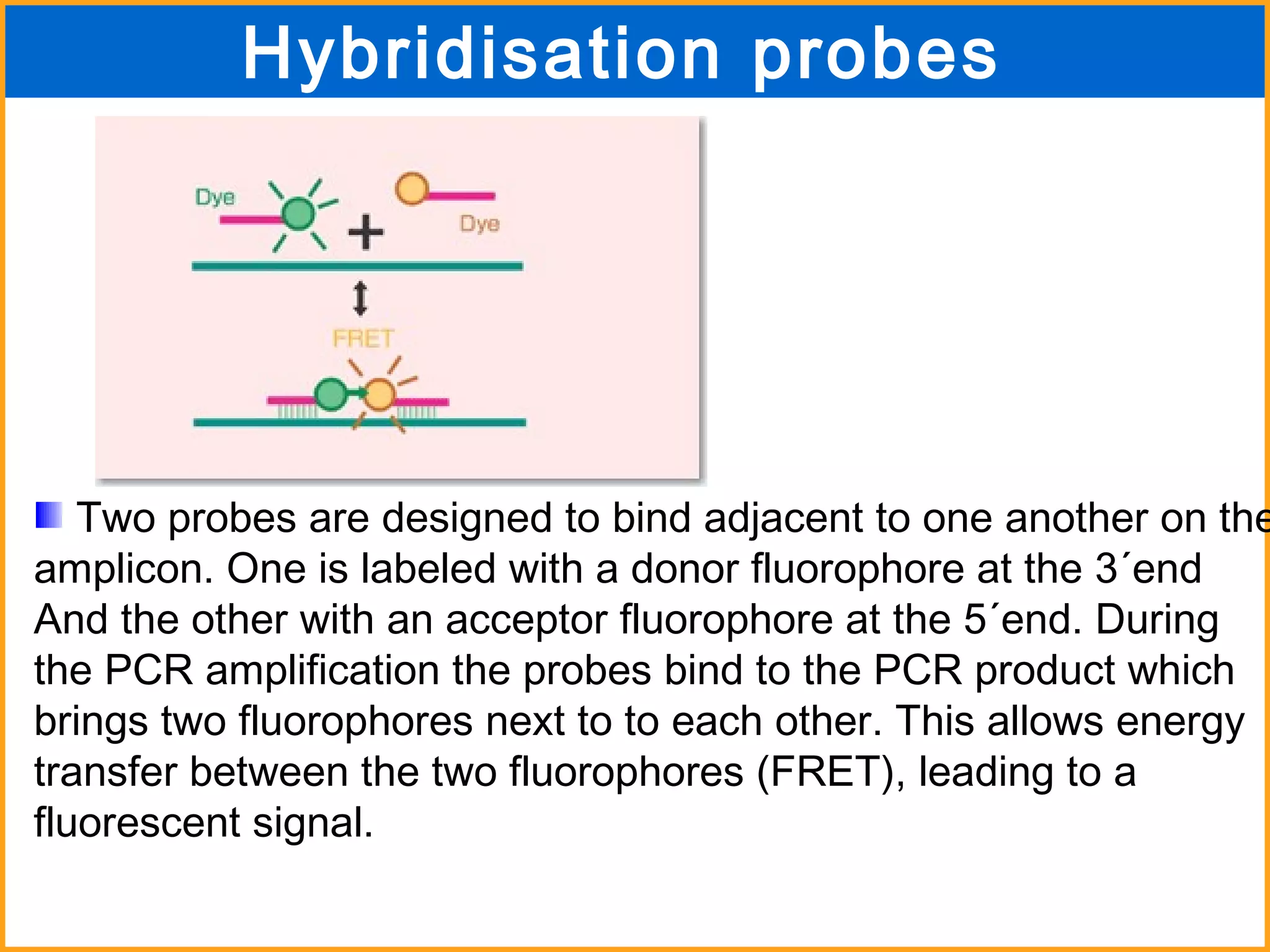
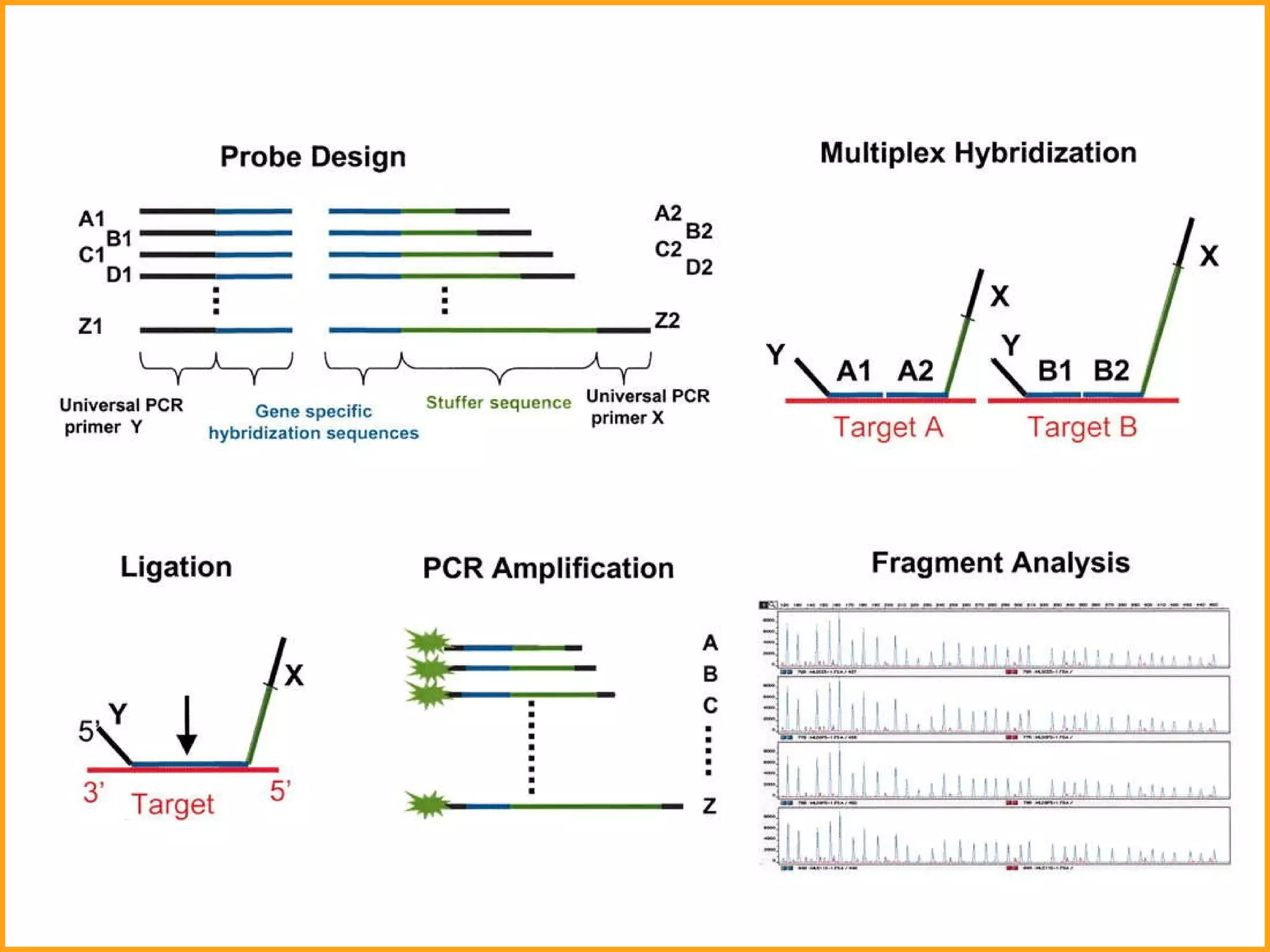
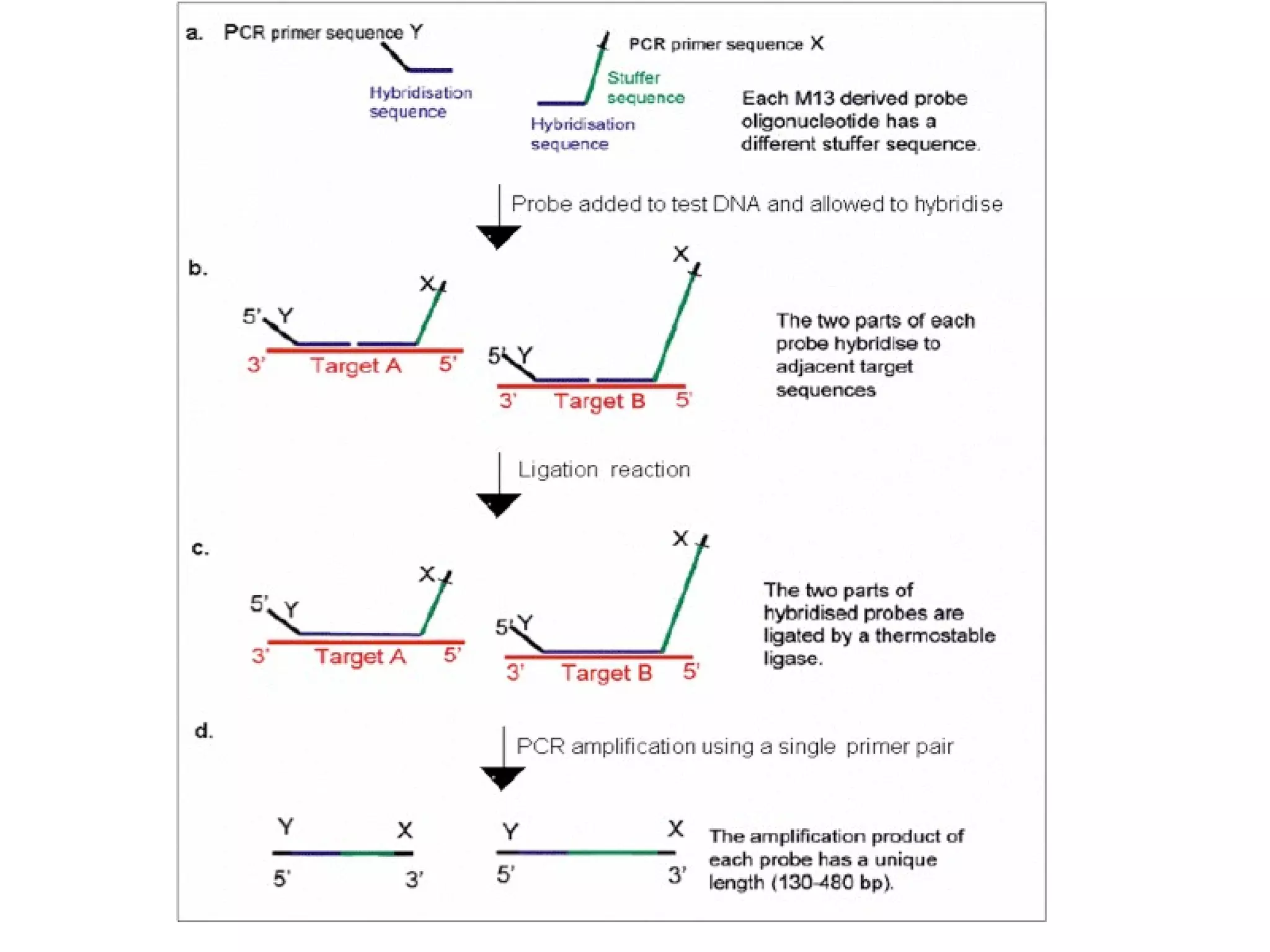
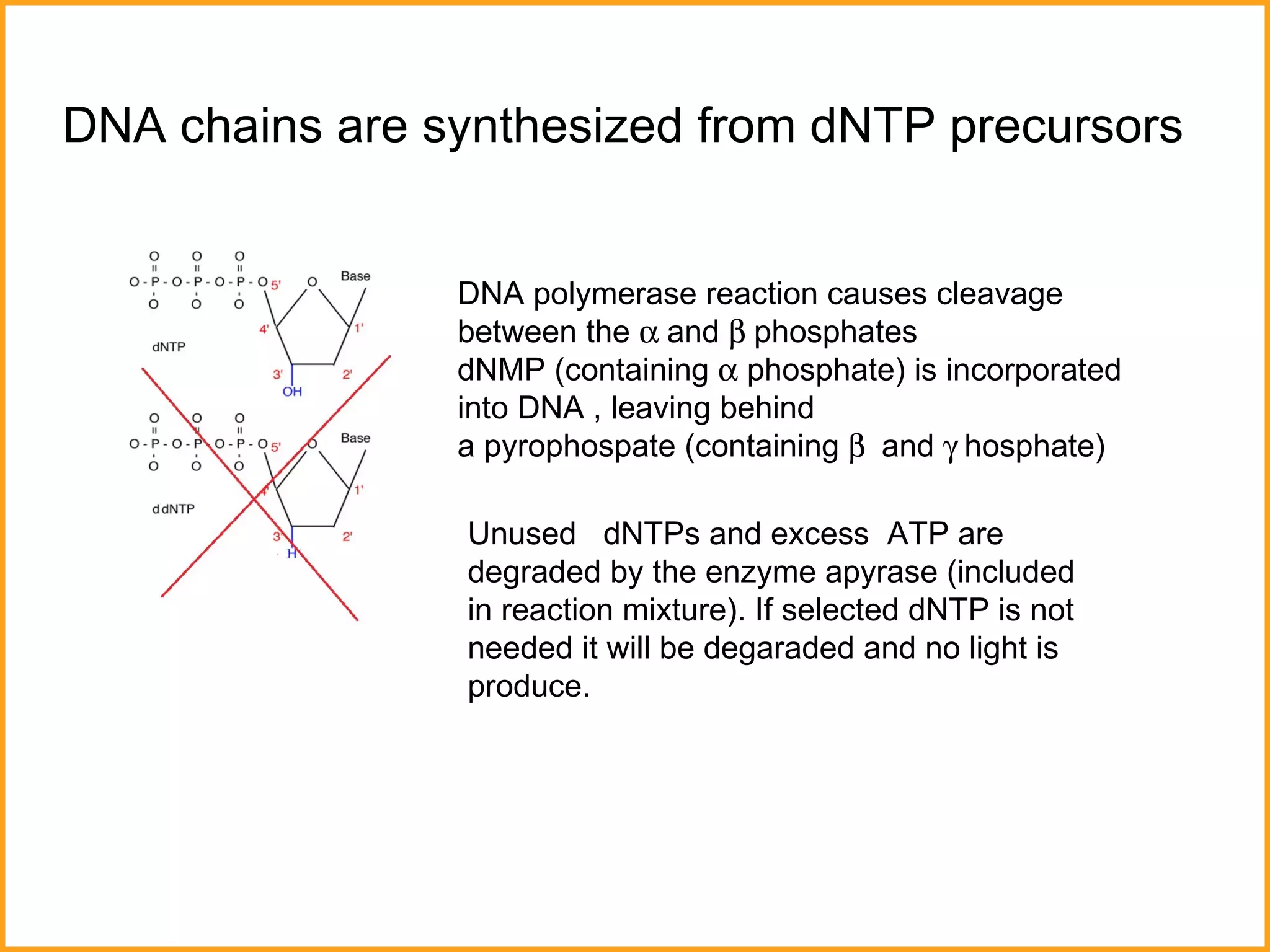
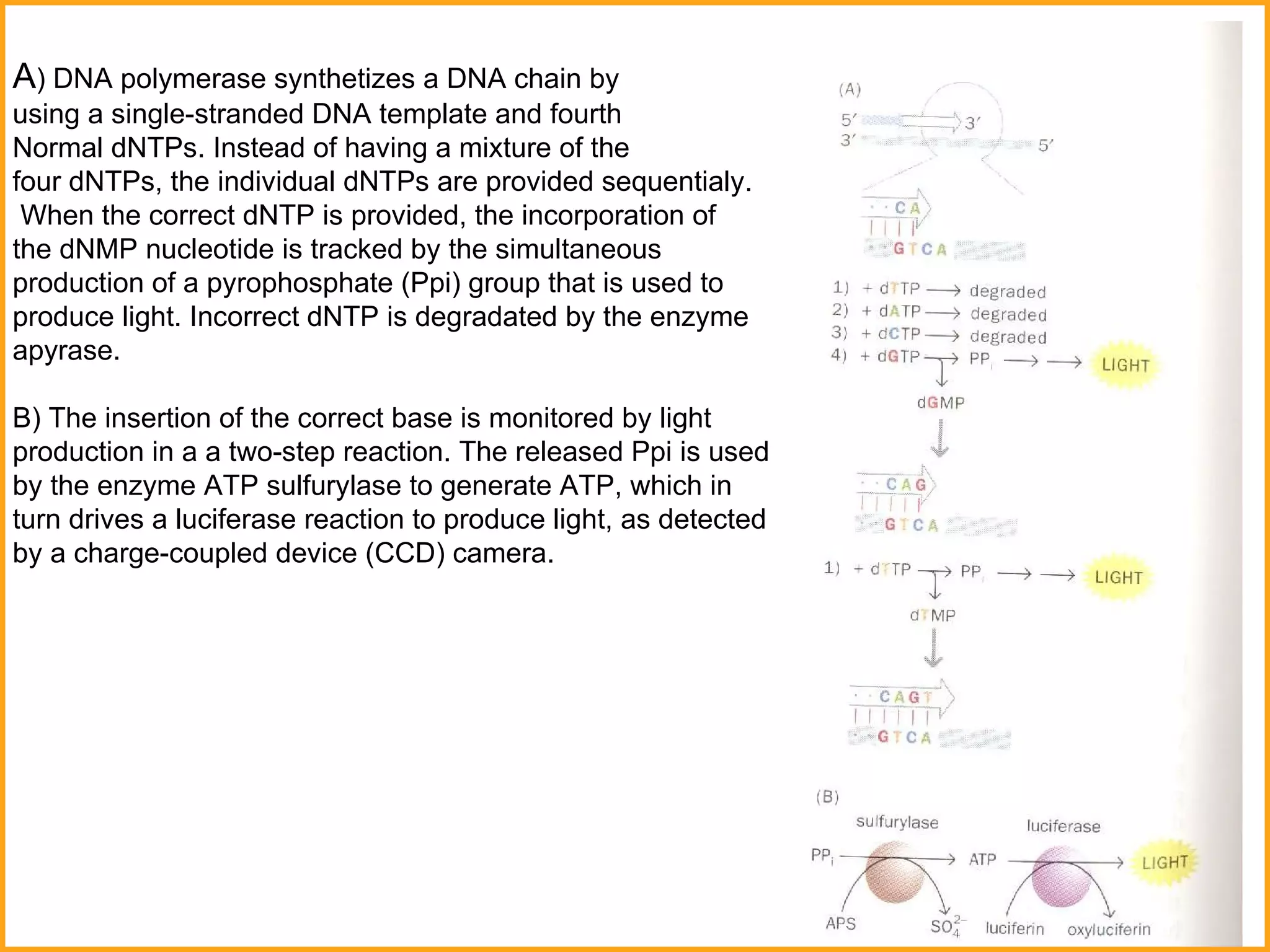
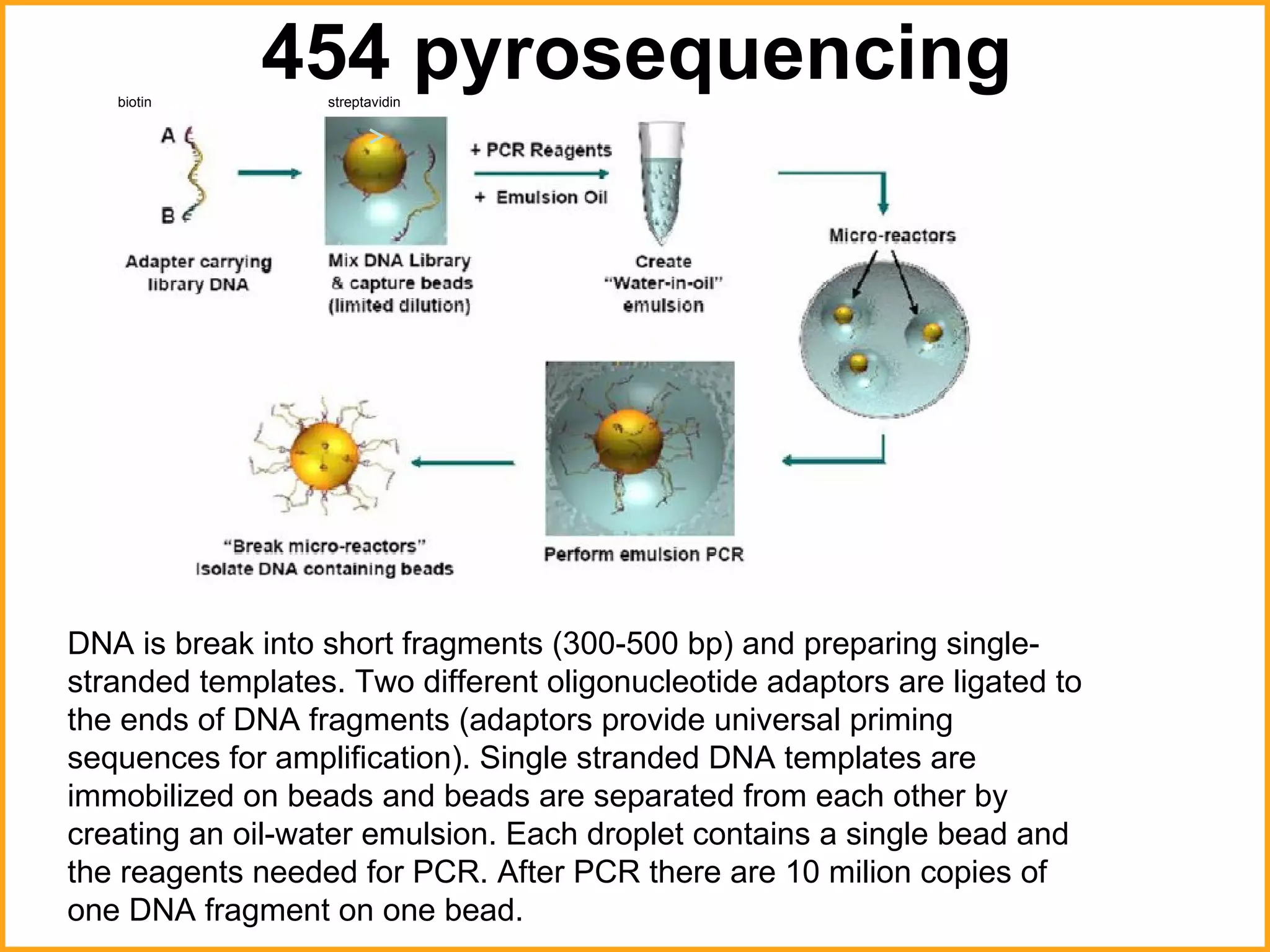
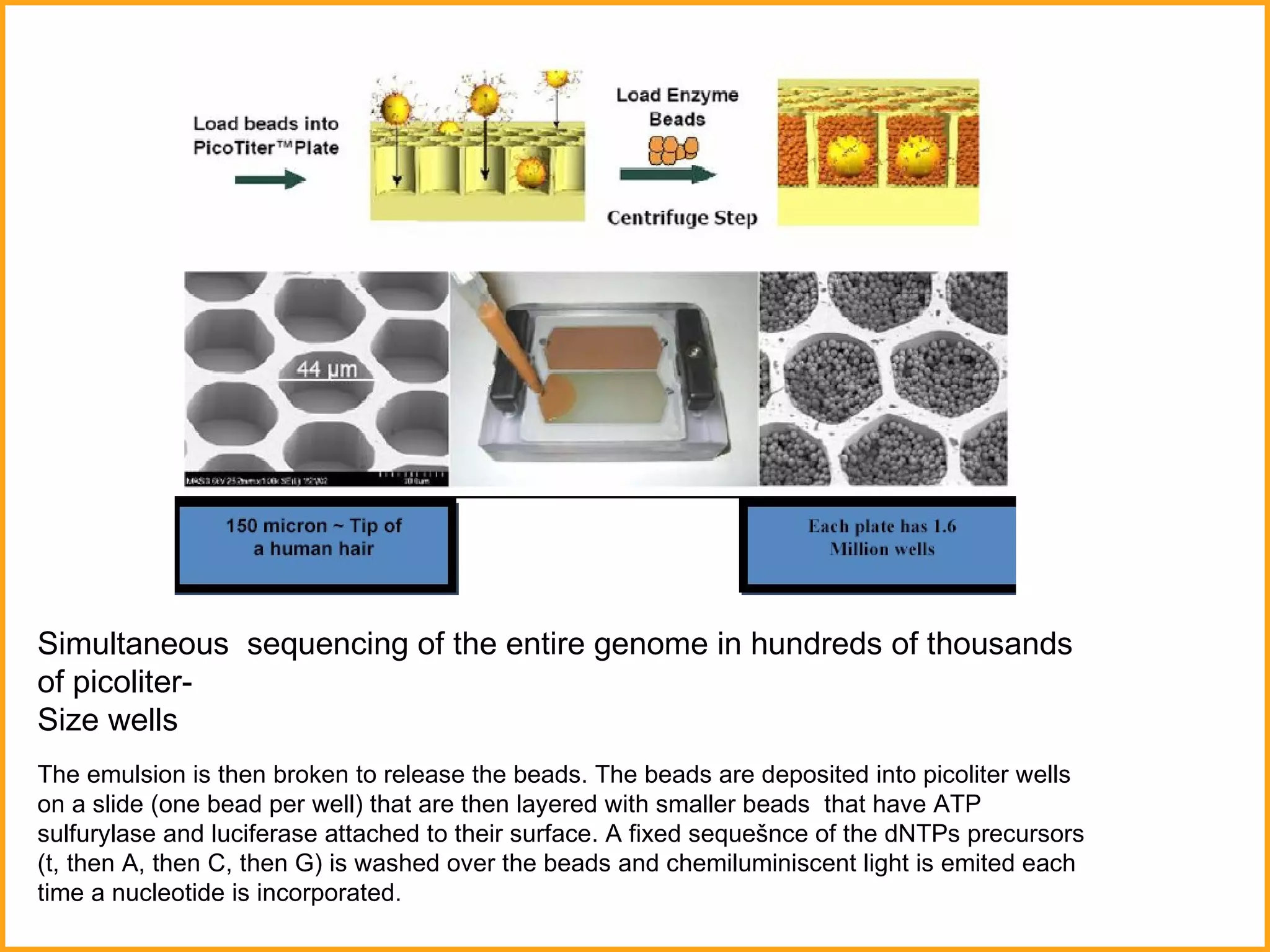
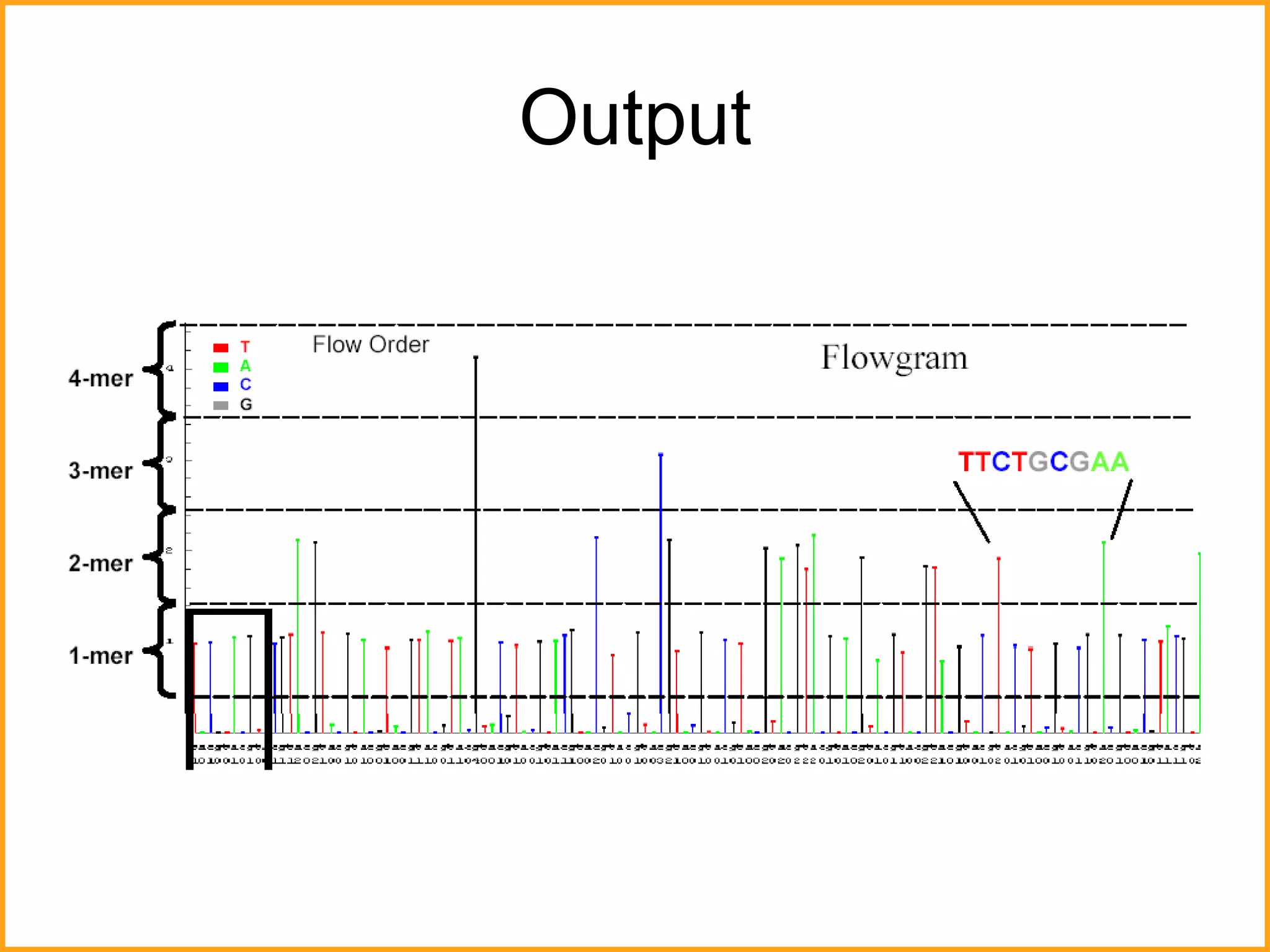
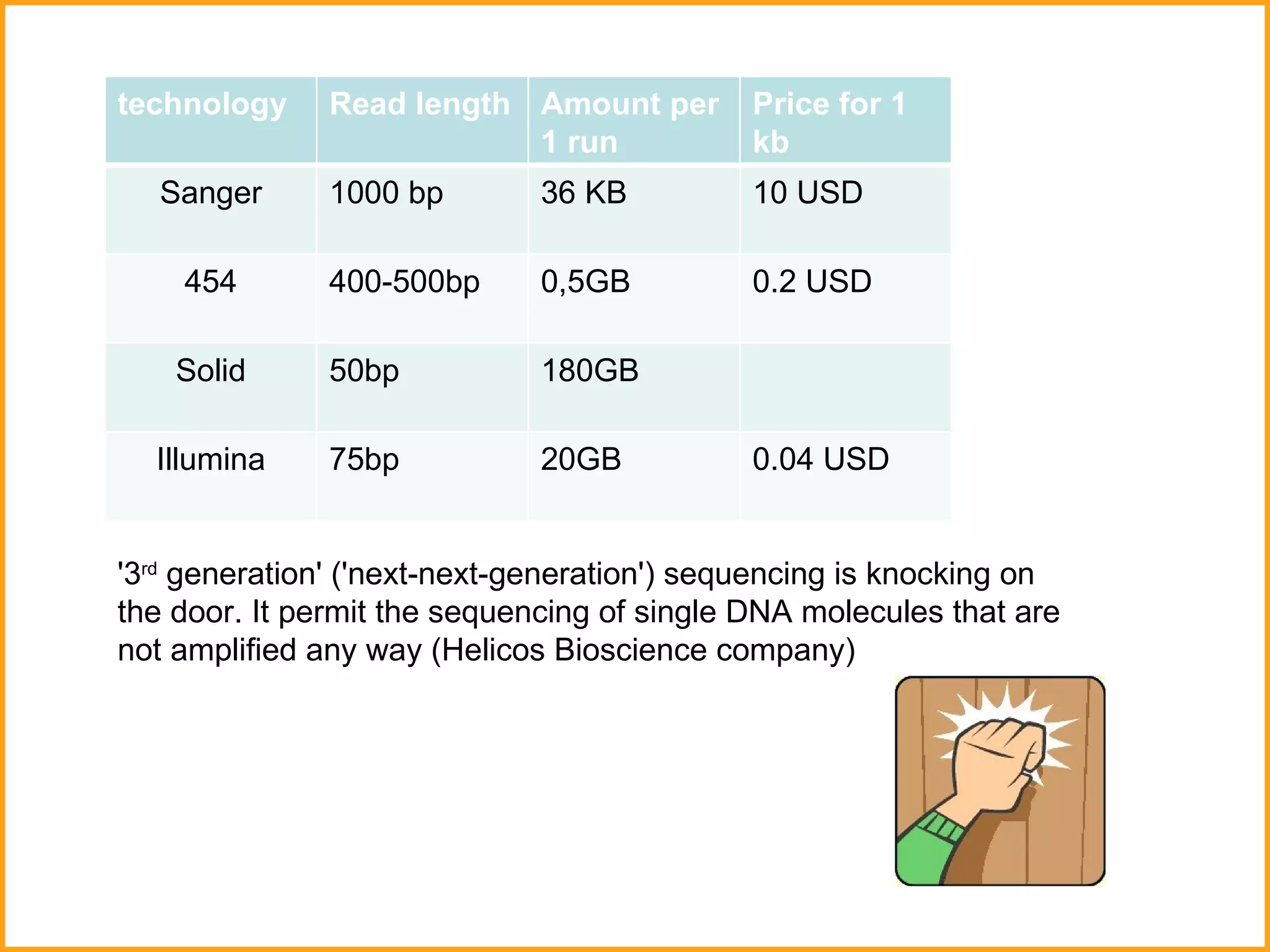
The document discusses various methods in molecular biology, including nucleic acid hybridization, DNA sequencing, real-time PCR, and DNA microarrays. Nucleic acid hybridization uses complementary base pairing between DNA or RNA probes and targets. DNA sequencing determines the nucleotide order using chain-terminating dideoxynucleotides. Real-time PCR quantifies DNA or RNA targets in real time using fluorescent probes. DNA microarrays allow analysis of gene expression patterns across thousands of genes.






























































![[Medicina] genetica molecular biology of the gene - watson j d , et al (5th e...](https://cdn.slidesharecdn.com/ss_thumbnails/8ounzyuirsojxks4isn9-signature-6ed96f61ba451dd55dc51bcf3f726eb18253d00f9f8650f3e5c1a1a36c1f18b2-poli-140721000012-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




































































