Download as PDF, PPTX




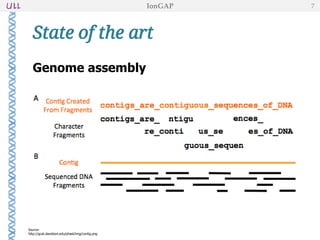

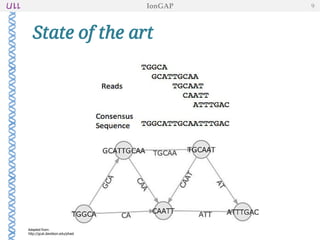

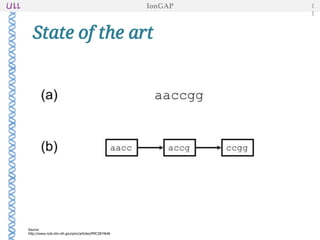
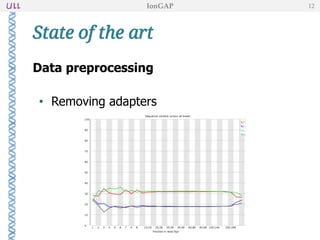
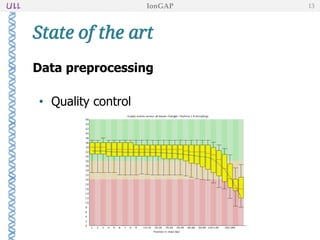

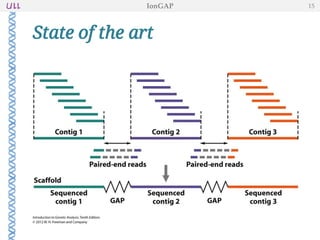



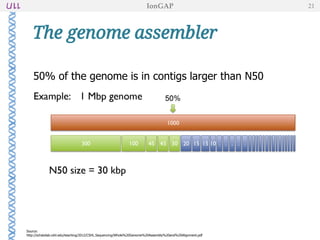
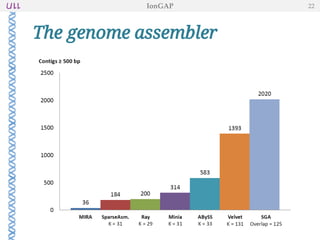
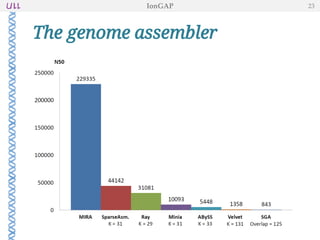

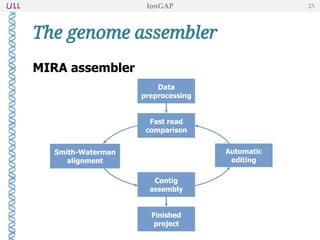

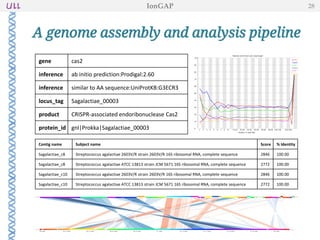

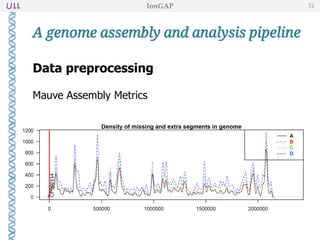



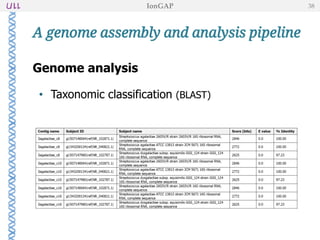


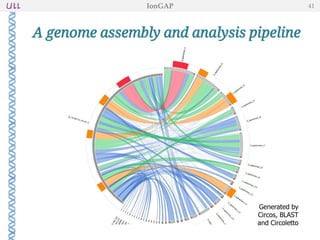
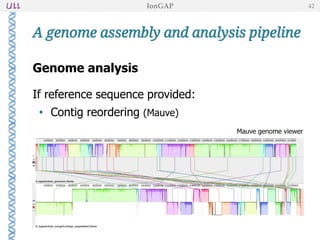
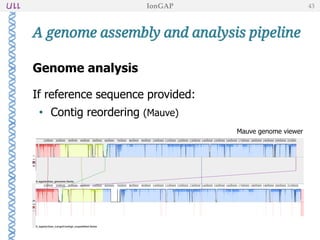


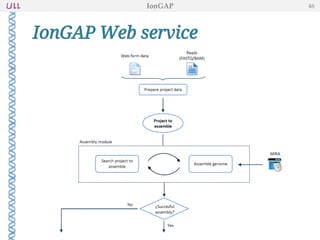
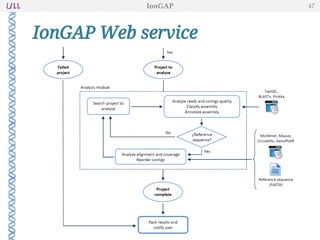

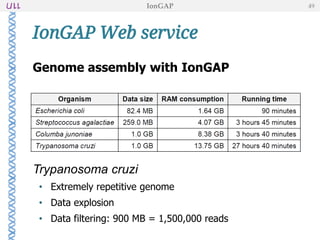

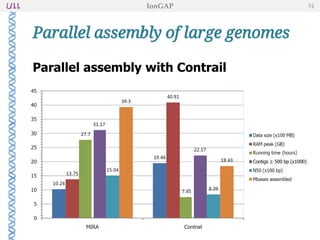





The document outlines the development of IonGap, an integrated software platform for the de novo assembly of genomic data derived from Ion Torrent sequencing. It includes a comprehensive analysis of available genome assemblers, concluding that the Mira assembler is most suitable for long-read assembly, while explaining various stages of genome processing and analysis. Future work includes options for cloud-based assembly and enhanced parallel processing for large genomes.






















































































