Downloaded 911 times



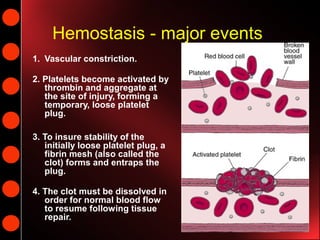















































Hemostasis is the process of blood clotting and subsequent dissolution of the clot following tissue repair. It involves 3 main events: 1) vascular constriction, 2) platelet aggregation forming a temporary plug, and 3) fibrin formation creating a stable clot. The clot must later dissolve for normal blood flow to resume. Von Willebrand disease is a bleeding disorder caused by deficient or abnormal von Willebrand factor, which helps platelets bind to collagen. It ranges from mild to severe and is diagnosed based on bleeding history and lab tests of von Willebrand factor and coagulation factors. Treatment depends on severity but may include desmopressin or coagulation factor replacement.























































































