Downloaded 136 times

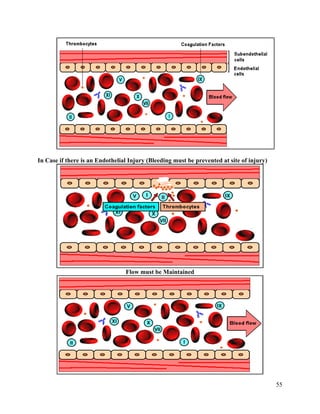
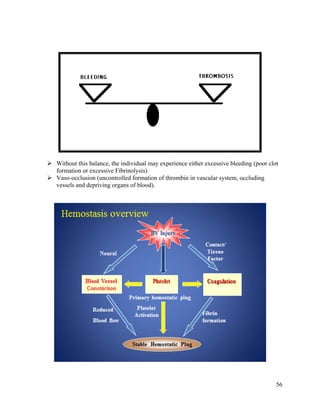




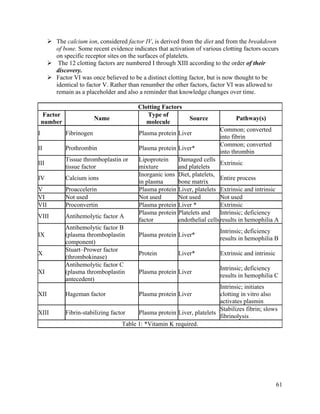
















This document summarizes hemostasis, the process by which bleeding is stopped. It discusses the three components of hemostasis - extravascular, vascular, and intravascular. The normal hemostasis process involves platelet plug formation and fibrin clot formation via the coagulation cascade. Coagulation factors, platelets, and fibrinogen are involved. Hemostasis is balanced by natural anticoagulants. Genetic or acquired bleeding disorders can result from deficiencies in specific coagulation factors or platelets. Common disorders discussed include hemophilia A/B/C and von Willebrand disease.























































































