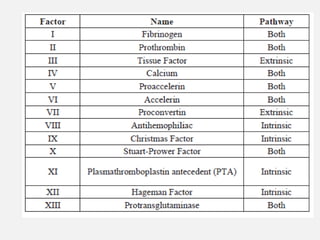
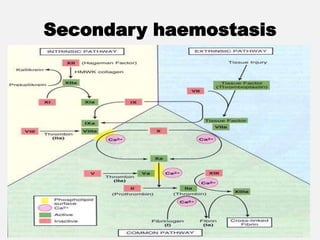
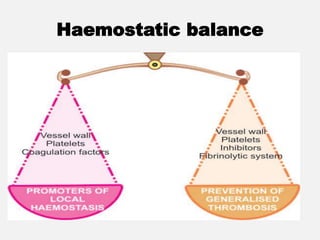
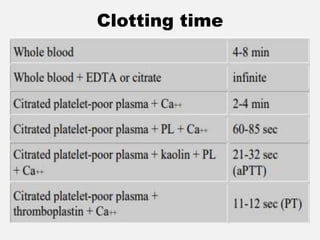
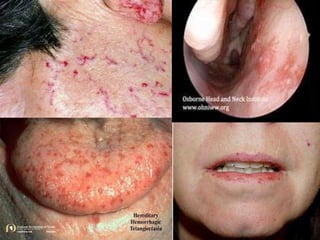
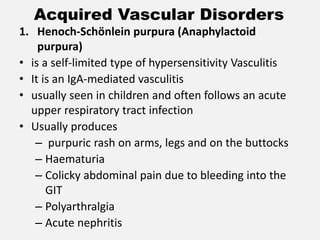
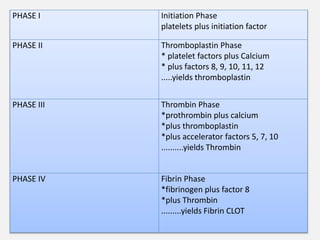
The document provides an extensive overview of haemostasis, detailing its mechanisms, phases, and the balance between coagulation and fibrinolysis. It discusses various disorders related to bleeding and clotting, including conditions such as haemophilia A and B, von Willebrand disease, and the impact of vitamin K deficiency. Additionally, it covers diagnostic tests, clinical features of coagulation disorders, and management strategies.



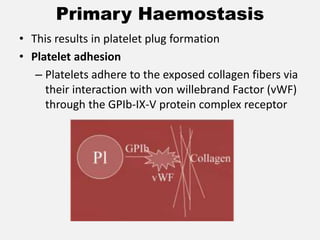
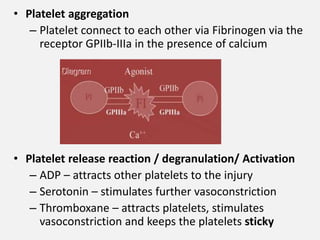
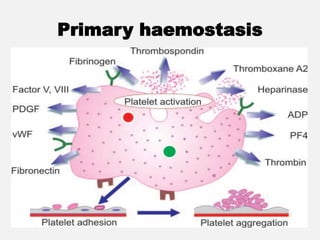
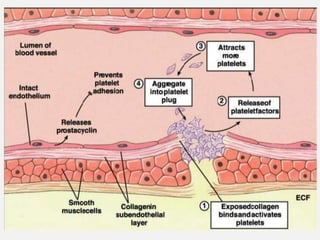













































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)







