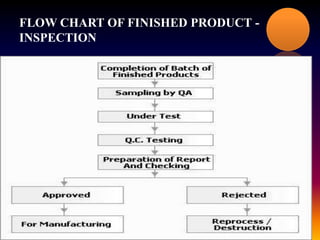
The document outlines the processes and guidelines for finished product release in the pharmaceutical industry, emphasizing the importance of quality review, quality audits, and compliance with WHO guidelines. It details standard operating procedures for batch release, sampling techniques, and the significance of auditing practices to ensure product safety and compliance. Conclusively, it highlights that quality reviews are essential for assessing the entire production process before market release.











































































![supriya.k_ppt[1].pptx documentation for QA and QC](https://cdn.slidesharecdn.com/ss_thumbnails/supriya-250809112814-eb4e6f1f-thumbnail.jpg?width=640&height=640&fit=bounds)

































