Downloaded 24 times
























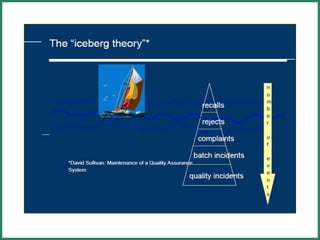





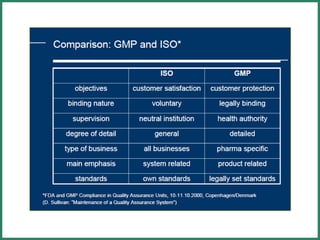
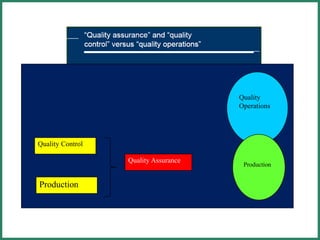


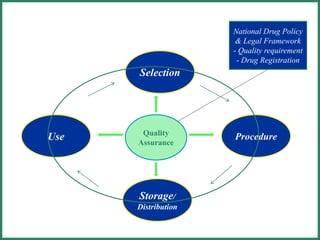

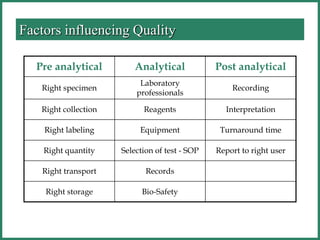



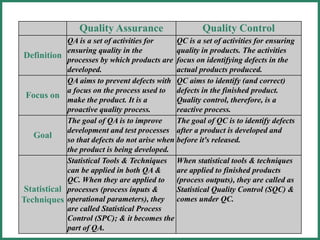



The document outlines Good Manufacturing Practice (GMP) regulations to ensure the safety, quality, and efficacy of pharmaceutical products through established standards and methods. It emphasizes the importance of quality assurance and control systems, documentation, and deviation investigations, while defining roles and responsibilities within quality control units. Additionally, the need for regular audits, compliance with regulatory requirements, and a risk-based approach to manufacturing is discussed to prevent contamination and ensure product integrity.


















































































