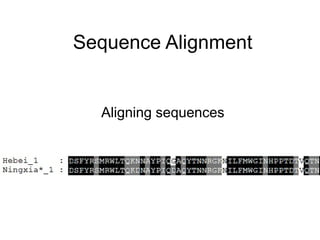
Sequence alignment
Alignment: Comparingtwo (pairwise)
or more (multiple) sequences.
Searching for a series of identical or
similar characters in the sequences.
5.
Why align?
VLSPAVKWAKV
||| ||||||
VLSEAVLWAKV
1.To detect if two sequences are homologous. If so,
homology may indicate similarity in structure (and
function).
2.Given a sequenced DNA, from an unknown region,
align it to the genome.
3.Required for evolutionary studies (e.g., tree
reconstruction).
4.To detect conservation (e.g., a tyrosine that is
evolutionary conserved is more likely to be a
phosphorylation site).
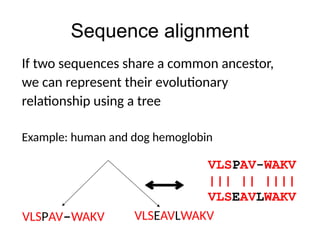
Sequence alignment
If twosequences share a common ancestor,
we can represent their evolutionary
relationship using a tree
Example: human and dog hemoglobin
VLSPAV-WAKV
||| || ||||
VLSEAVLWAKV
VLSPAV-WAKV VLSEAVLWAKV
13.
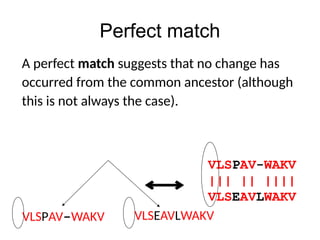
Perfect match
VLSPAV-WAKV
||| ||||||
VLSEAVLWAKV
VLSPAV-WAKV VLSEAVLWAKV
A perfect match suggests that no change has
occurred from the common ancestor (although
this is not always the case).
14.
A substitution
VLSPAV-WAKV
||| ||||||
VLSEAVLWAKV
VLSPAV-WAKV VLSEAVLWAKV
A substitution suggests that at least one
change has occurred since the common
ancestor
We cannot say in which lineage it occurred!
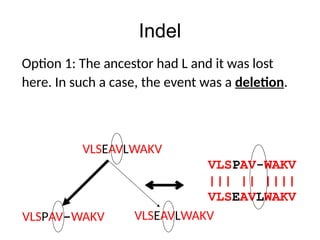
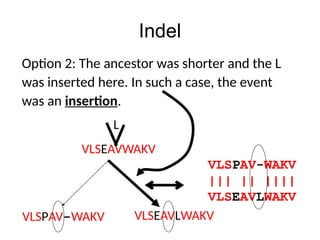
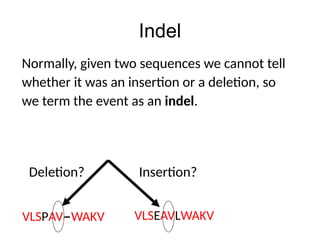
Indel
VLSPAV-WAKV
Normally, given twosequences we cannot tell
whether it was an insertion or a deletion, so
we term the event as an indel.
VLSEAVLWAKV
Deletion? Insertion?
18.
Indels in proteincoding genes
Indels in protein coding genes are often of 3bp,
6bp, 9bp, etc...
Why?
Gene Search
In fact, searching for indels of length 3K
(K=1,2,3,…) can help algorithms that search a
genome for coding regions
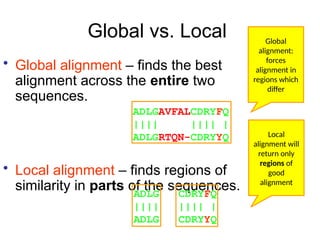
Global vs. Local
•Global alignment – finds the best
alignment across the entire two
sequences.
• Local alignment – finds regions of
similarity in parts of the sequences.
ADLGAVFALCDRYFQ
|||| |||| |
ADLGRTQN-CDRYYQ
ADLG CDRYFQ
|||| |||| |
ADLG CDRYYQ
Global
alignment:
forces
alignment in
regions which
differ
Local
alignment will
return only
regions of
good
alignment
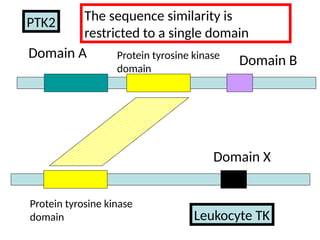
Proteins are comprisedof domains
Domain B
Protein tyrosine
kinase domain
Domain A
Human PTK2 :
Global alignment may be problematic!
23.
Domain X
Protein tyrosinekinase
domain
Domain B
Protein tyrosine kinase
domain
Domain A
Leukocyte TK
PTK2 The sequence similarity is
restricted to a single domain
Conclusions
Use global alignmentwhen the two sequences
share the same overall sequence arrangement.
Use local alignment to detect regions of
similarity.
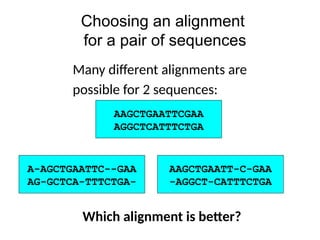
Choosing an alignment
fora pair of sequences
AAGCTGAATTCGAA
AGGCTCATTTCTGA
AAGCTGAATT-C-GAA
AGGCT-CATTTCTGA
-
A-AGCTGAATTC--GAA
AG-GCTCA-TTTCTGA-
Which alignment is better?
Many different alignments are
possible for 2 sequences:
Alignment scoring -scoring of
sequence similarity:
Assumes independence between positions:
each position is considered separately
Scores each position:
• Positive if identical (match)
• Negative if different (mismatch or gap)
Total score = sum of position scores
Can be positive or negative
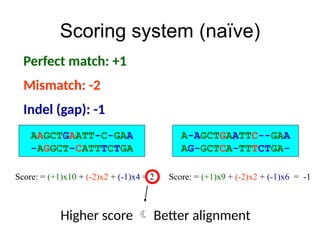
Scoring system
• Inthe example above, the choice of +1 for
match,-2 for mismatch, and -1 for gap is
quite arbitrary
• Different scoring systems different
alignments
• We want a good scoring system…
35.
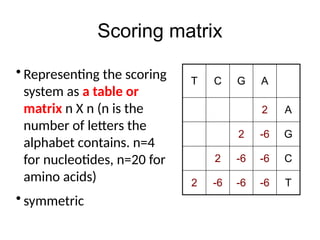
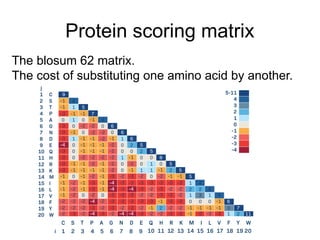
Scoring matrix
T CG A
2 A
2 -6 G
2 -6 -6 C
2 -6 -6 -6 T
• Representing the scoring
system as a table or
matrix n X n (n is the
number of letters the
alphabet contains. n=4
for nucleotides, n=20 for
amino acids)
• symmetric
36.
DNA scoring matrices
•Uniform substitutions between all nucleotides:
T C G A From
To
-6 -6 -6 2 A
-6 -6 2 -6 G
-6 2 -6 -6 C
2 -6 -6 -6 T
Match
Mismatch
37.
DNA scoring matrices
Cantake into account biological phenomena
such as:
• Transition-transversion
38.
DNA scoring matrices
•Non-uniform substitutions between all nucleotides:
T C G A From
To
-6 -6 -2 2 A
-6 -6 2 -2 G
-2 2 -6 -6 C
2 -2 -6 -6 T
Match
Mismatch
39.
Scoring gaps (I)
Inadvanced algorithms, two gaps of one amino-
acid are given a different score than one gap of
two amino acids. This is solved by giving a penalty
to each gap that is opened.
Gap extension penalty < Gap opening penalty
40.
Scoring gaps (II)
Assumethat the gap opening cost is -3. The
length contributes -1 per base pair
AGGGTTC—GA
AGGGTTCTGA
Score = -4
AGGGTT-—GA
AGGGTTCTGA
Score = -5
AGGGT--—GA
AGGGTTCTGA
Score = -6
AGGG--C—GA
AGGGTTCTGA
Score = -9
Linear penalty
Optimal alignment algorithms
•Needleman-Wunsch (global) [1970]
• Smith-Waterman (local) [1981]
Only length(seq1) x length(seq2) operations!
Length of seq1,seq2 #
operations Instead of
10 100 1,000,000
20 400 100,000,000,000,000
30 900 10,000,000,000,000,000,000,
000
45.
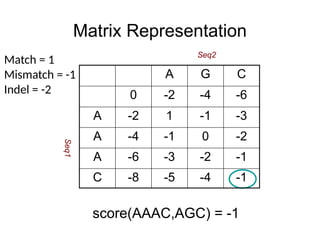
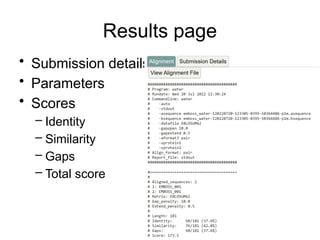
Matrix Representation
Match =1
Mismatch = -1
Indel = -2
A G C
0 -2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
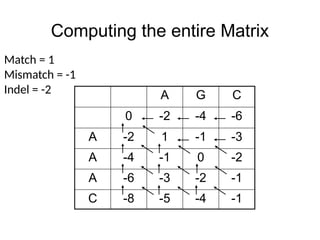
score(AAAC,AGC) = -1
Seq2
Seq1
46.
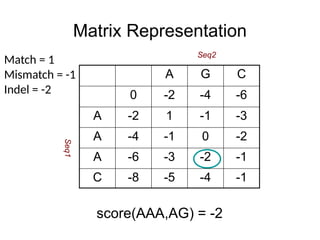
Matrix Representation
score(AAA,AG) =-2
A G C
0 -2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
Match = 1
Mismatch = -1
Indel = -2
Seq2
Seq1
47.
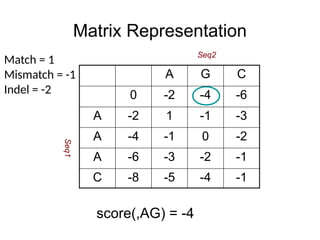
Matrix Representation
score(,AG) =-4
A G C
0 -2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
Match = 1
Mismatch = -1
Indel = -2
Seq2
Seq1
48.
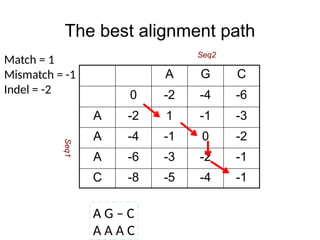
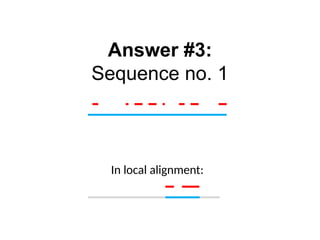
The best alignmentpath
A G – C
A A A C
Match = 1
Mismatch = -1
Indel = -2
Seq2
Seq1
A G C
0 -2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
49.
Mi,j = MAXIMUM[
Mi-1,j-1 + Si,j (match/mismatch in the diagonal),
Mi,j-1 + w (gap in sequence #1),
Mi-1,j + w (gap in sequence #2)]
Computing the score of each cell
Mi,j = the score in cell (i,j)
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
50.
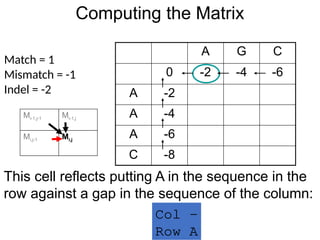
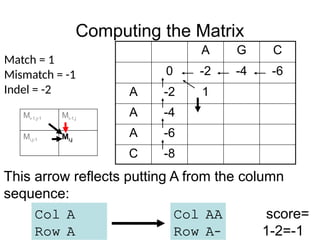
Computing the Matrix
AG C
0 -2 -4 -6
A -2
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
This cell reflects putting A in the sequence in the
row against a gap in the sequence of the column:
Col -
Row A
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
51.
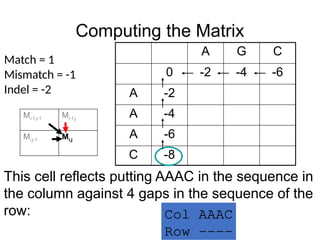
Computing the Matrix
AG C
0 -2 -4 -6
A -2
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
This cell reflects putting AAAC in the sequence in
the column against 4 gaps in the sequence of the
row: Col AAAC
Row ----
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
52.
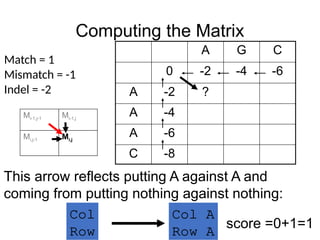
Computing the Matrix
AG C
0 -2 -4 -6
A -2 ?
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
This arrow reflects putting A against A and
coming from putting nothing against nothing:
Col A
Row A
Col
Row
score =0+1=1
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
53.
Computing the Matrix
AG C
0 -2 -4 -6
A -2 ?
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
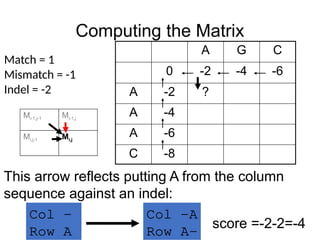
This arrow reflects putting A from the column
sequence against an indel:
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
Col -A
Row A-
Col -
Row A
score =-2-2=-4
54.
Computing the Matrix
AG C
0 -2 -4 -6
A -2 ?
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
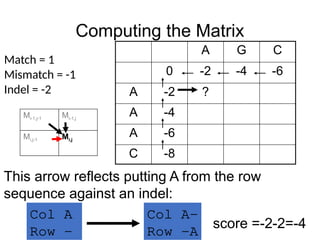
This arrow reflects putting A from the row
sequence against an indel:
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
Col A-
Row -A
Col A
Row -
score =-2-2=-4
55.
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
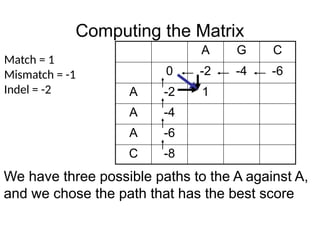
We have three possible paths to the A against A,
and we chose the path that has the best score
56.
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
We have three possible paths to the A against A,
and we chose the path that has the best score
57.
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
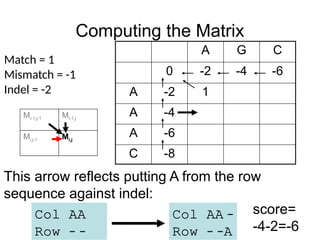
This arrow reflects putting A from the row
sequence against indel:
Col AA –
Row – –A
Col AA
Row – –
score=
-4-2=-6
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
58.
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
This arrow reflects putting A from the column
sequence:
Col AA
Row A–
Col A
Row A
score=
1-2=-1
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
59.
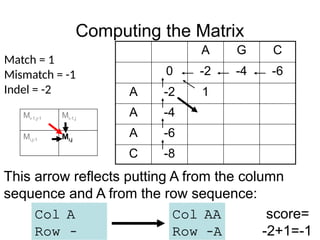
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
This arrow reflects putting A from the column
sequence and A from the row sequence:
Col AA
Row –A
Col A
Row –
score=
-2+1=-1
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j
60.
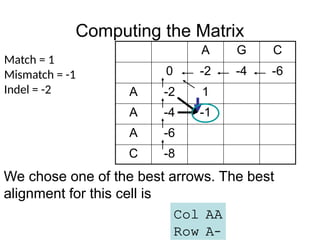
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4 -1
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
We chose one of the best arrows. The best
alignment for this cell is
Col AA
Row A–
61.
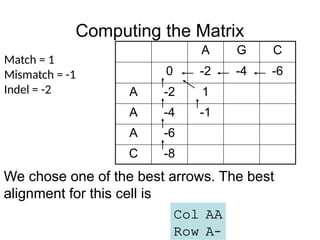
Computing the Matrix
AG C
0 -2 -4 -6
A -2 1
A -4 -1
A -6
C -8
Match = 1
Mismatch = -1
Indel = -2
We chose one of the best arrows. The best
alignment for this cell is
Col AA
Row A–
62.
Computing the entireMatrix
A G C
0 -2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
Match = 1
Mismatch = -1
Indel = -2
63.
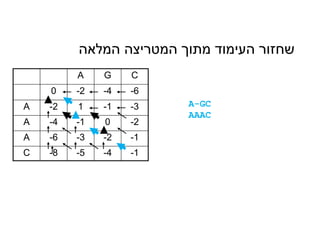
A G C
0-2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
המלאה המטריצה מתוך העימוד שחזור
AG-C
AAAC
We chose a path that goes from the end to
the beginning (marked here in Red). This is
called the Trace Back
64.
A G C
0-2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
המלאה המטריצה מתוך העימוד שחזור
A-GC
AAAC
65.
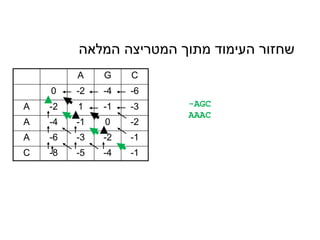
A G C
0-2 -4 -6
A -2 1 -1 -3
A -4 -1 0 -2
A -6 -3 -2 -1
C -8 -5 -4 -1
המלאה המטריצה מתוך העימוד שחזור
-AGC
AAAC
66.
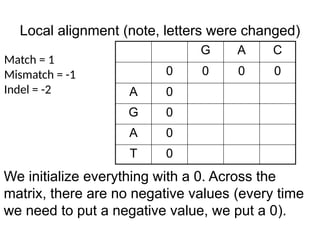
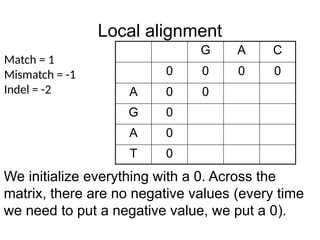
Local alignment (note,letters were changed)
G A C
0 0 0 0
A 0
G 0
A 0
T 0
Match = 1
Mismatch = -1
Indel = -2
We initialize everything with a 0. Across the
matrix, there are no negative values (every time
we need to put a negative value, we put a 0).
67.
Local alignment
G AC
0 0 0 0
A 0 0
G 0
A 0
T 0
Match = 1
Mismatch = -1
Indel = -2
We initialize everything with a 0. Across the
matrix, there are no negative values (every time
we need to put a negative value, we put a 0).
68.
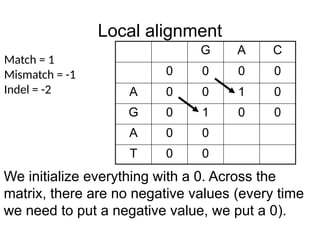
Local alignment
G AC
0 0 0 0
A 0 0 1 0
G 0 1 0 0
A 0 0
T 0 0
Match = 1
Mismatch = -1
Indel = -2
We initialize everything with a 0. Across the
matrix, there are no negative values (every time
we need to put a negative value, we put a 0).
69.
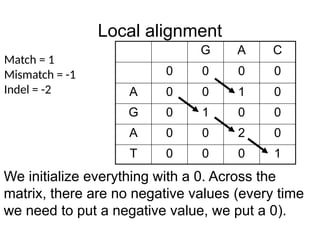
Local alignment
G AC
0 0 0 0
A 0 0 1 0
G 0 1 0 0
A 0 0 2 0
T 0 0 0 1
Match = 1
Mismatch = -1
Indel = -2
We initialize everything with a 0. Across the
matrix, there are no negative values (every time
we need to put a negative value, we put a 0).
70.
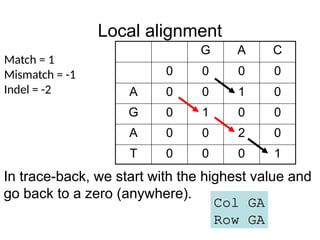
Local alignment
G AC
0 0 0 0
A 0 0 1 0
G 0 1 0 0
A 0 0 2 0
T 0 0 0 1
Match = 1
Mismatch = -1
Indel = -2
In trace-back, we start with the highest value and
go back to a zero (anywhere).
Col GA
Row GA
71.
Global and Localpairwise alignments -
summary
• Global alignment – finds the best
alignment over all the positions
• Local alignment – finds regions of
similarity in parts of the sequences
72.
Global & LocalAlignments
Global Most useful when the sequences are similar in the
entire alignment.
Local More useful for dissimilar sequences that
are suspected to contain regions of similarity
or similar sequence motifs.
73.
Pairwise alignment
• Localalignment – Waterman algorithm
• Global alignment – Needleman algorithm
• Protein and nucleotide alignments
• Input: two sequences
• Output: optimal sequence alignment
74.
Question #1:
For highlysimilar sequences, which method
is more compatible for alignment?
Global
Local
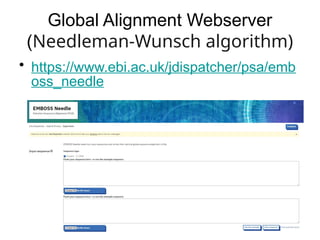
Local Alignment Webserver(Smith-
Waterman algorithm)
• https://www.ebi.ac.uk/jdispatcher/psa/emb
oss_water
78.
Altering parameters
• Scoringmatrix, e.g. BLOSUM:
– Higher numbers in matrix for similar
sequences
• Gap open & extend penalty
– Higher gap open penalty for similar
sequences
– If gap length matters increase gap extend
penalty
• Output format (pair)
BLOcks SUbstitution Matrix
•Based on comparisons of blocks of sequences from the
Blocks database.
• BLOSUM62 was built using data from sequences that
were up to 62% identical.



























![Optimal alignment algorithms
• Needleman-Wunsch (global) [1970]
• Smith-Waterman (local) [1981]
Only length(seq1) x length(seq2) operations!
Length of seq1,seq2 #
operations Instead of
10 100 1,000,000
20 400 100,000,000,000,000
30 900 10,000,000,000,000,000,000,
000](https://image.slidesharecdn.com/pairwisesequencealignment-250405162237-a67950f9/85/Pairwise-Sequence-Alignment-is-alignment-pptx-44-320.jpg)
![Mi,j = MAXIMUM[
Mi-1, j-1 + Si,j (match/mismatch in the diagonal),
Mi,j-1 + w (gap in sequence #1),
Mi-1,j + w (gap in sequence #2)]
Computing the score of each cell
Mi,j = the score in cell (i,j)
Mi-1,j-1 Mi-1,j
Mi,j-1 Mi,j](https://image.slidesharecdn.com/pairwisesequencealignment-250405162237-a67950f9/85/Pairwise-Sequence-Alignment-is-alignment-pptx-49-320.jpg)





























































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)
