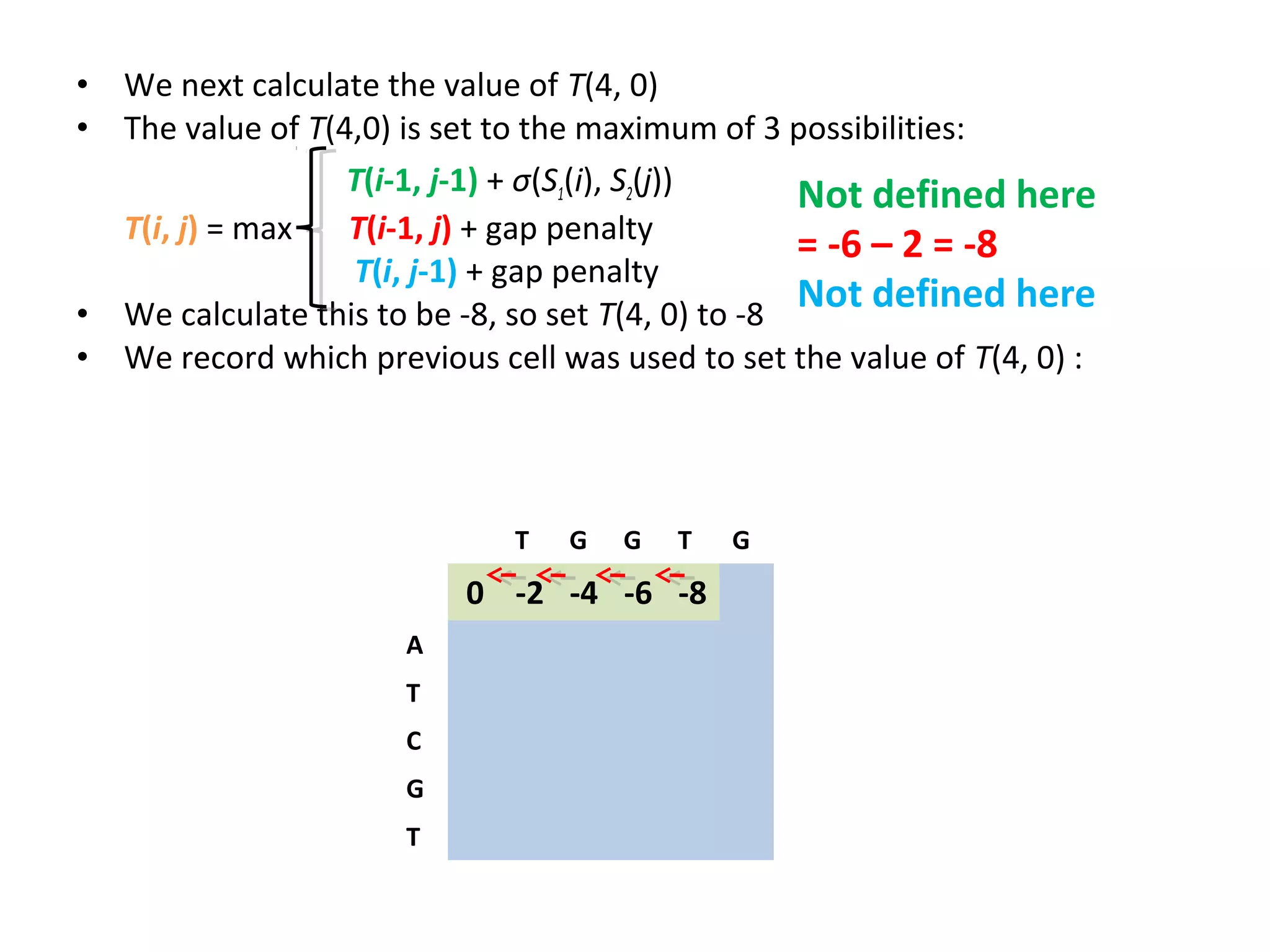
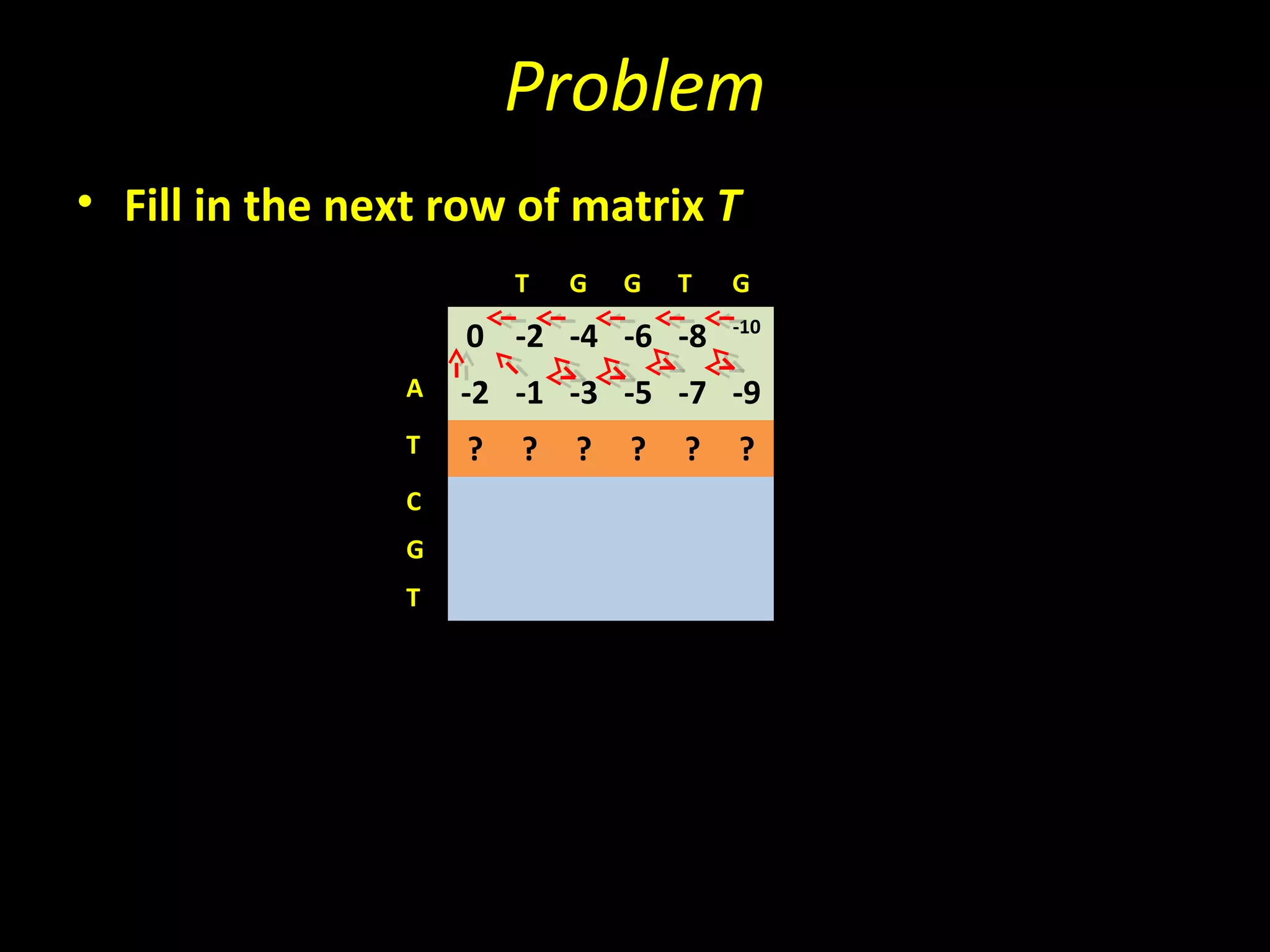
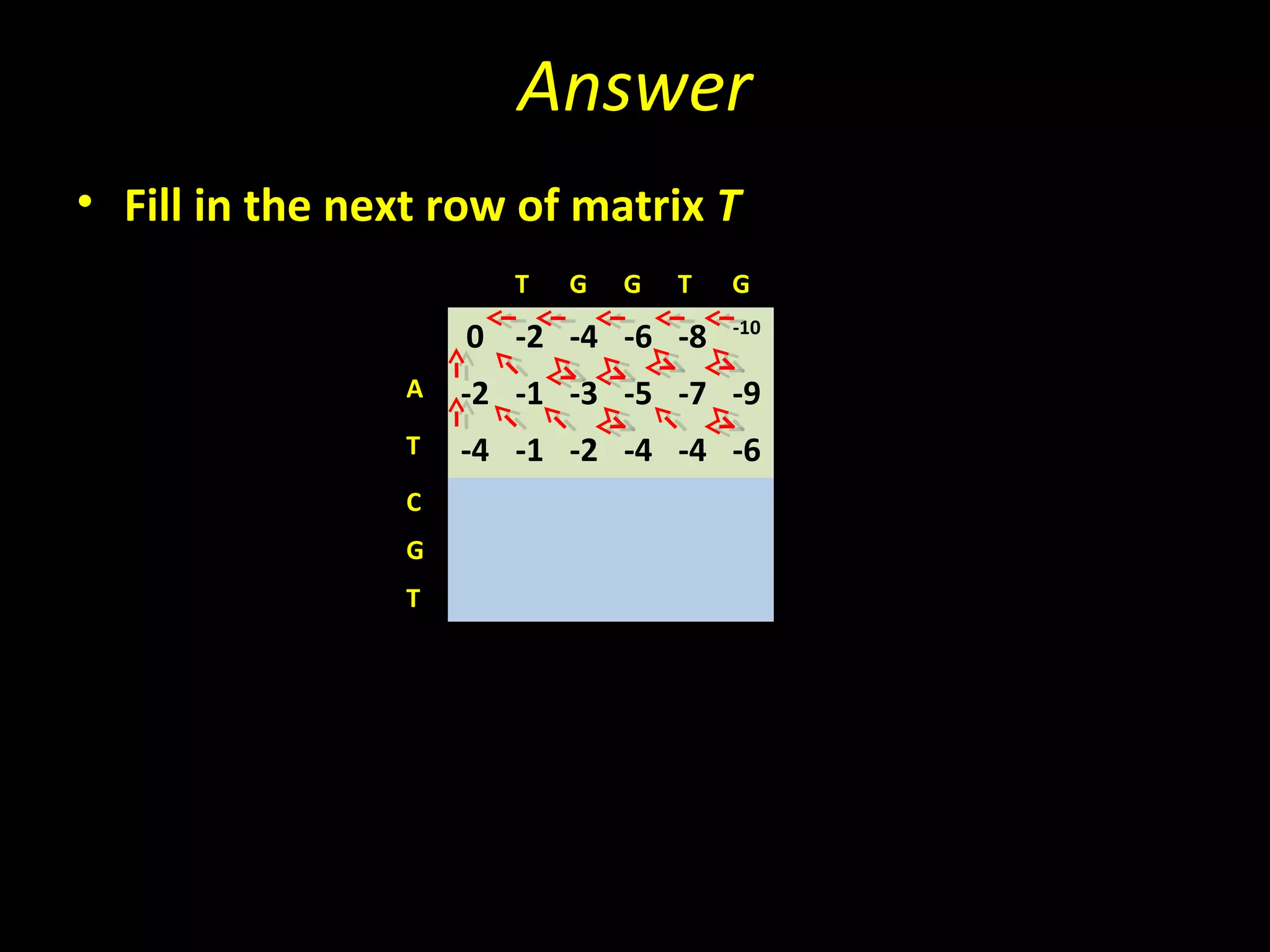
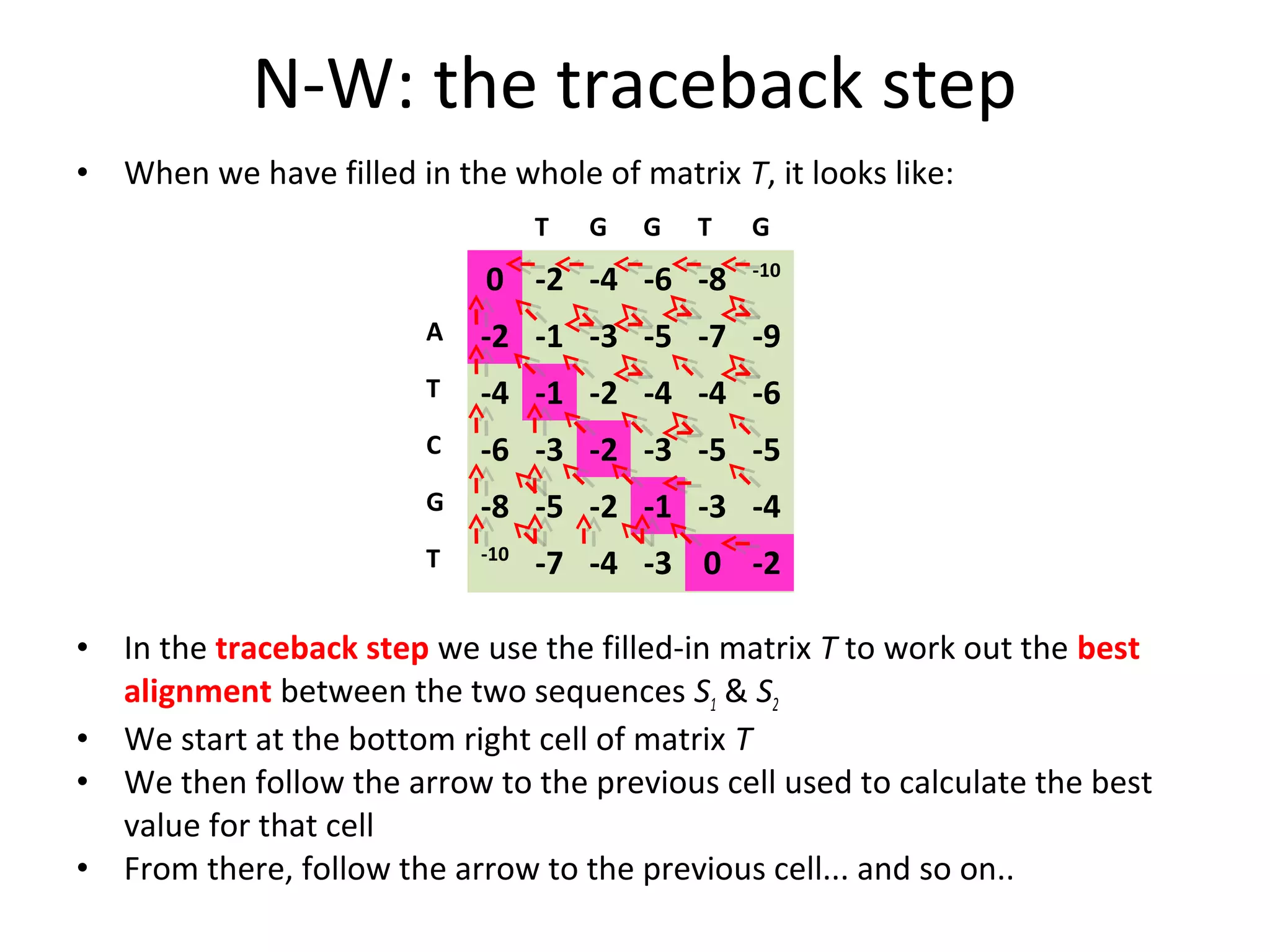
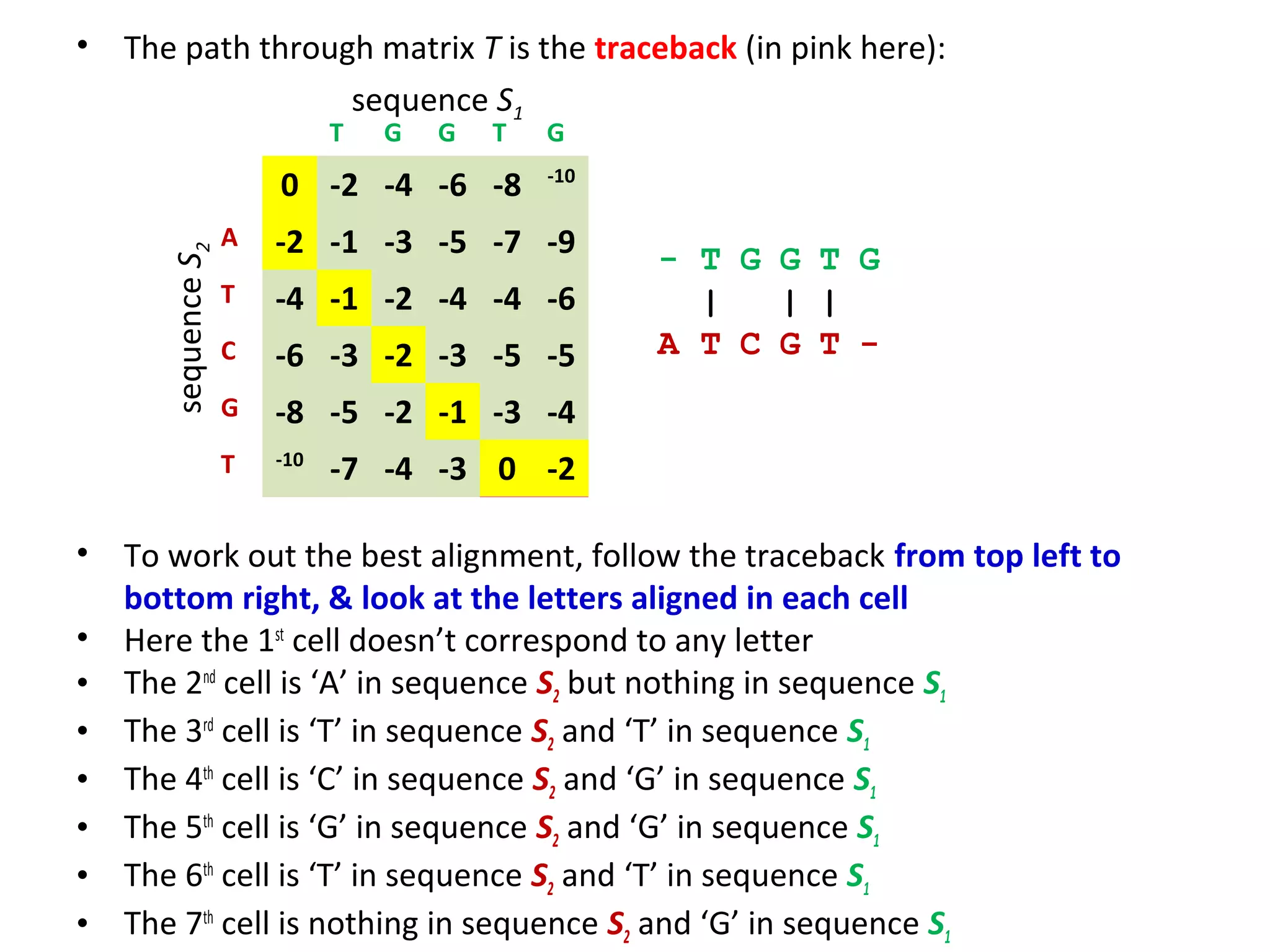
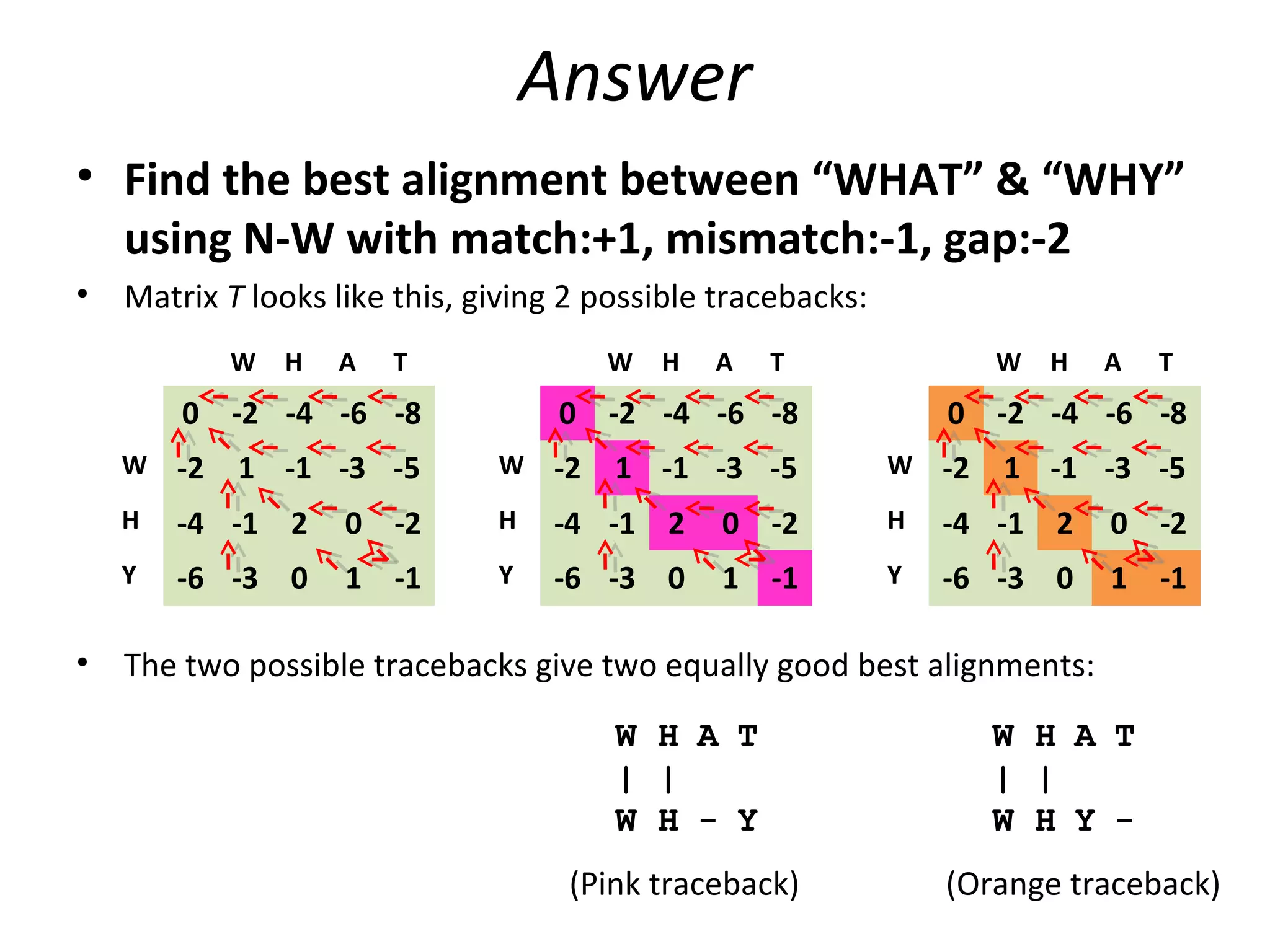
The Needleman-Wunsch algorithm finds the optimal global alignment of two nucleotide or protein sequences. It works by filling a matrix using a recursive formula that considers the best score from adjacent cells, incorporating substitution scores and gap penalties. This algorithm runs in quadratic time compared to assessing all possible alignments individually, which runs in exponential time. For two sequences of length n, the Needleman-Wunsch algorithm is much faster, taking n^2 time instead of the 2^n time needed to assess all alignments individually.