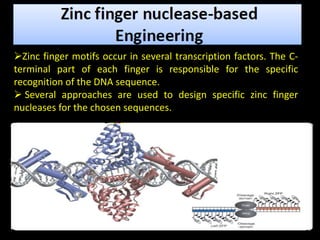
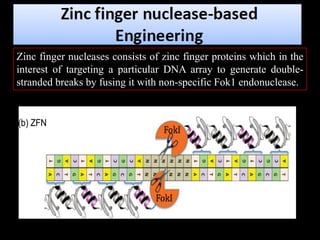
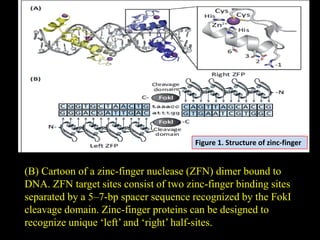
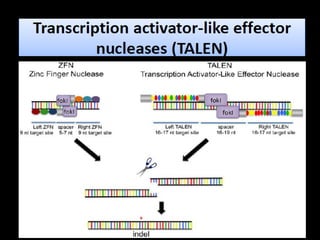
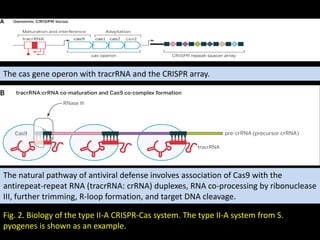
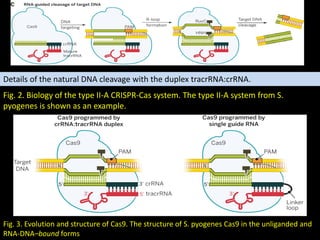
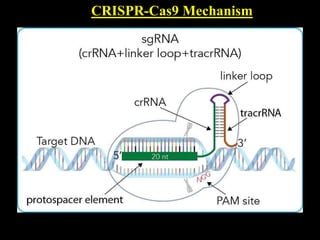
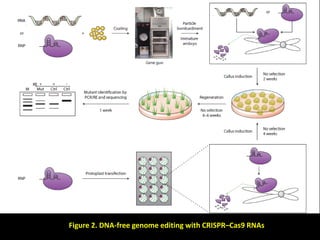
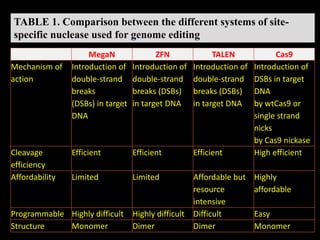
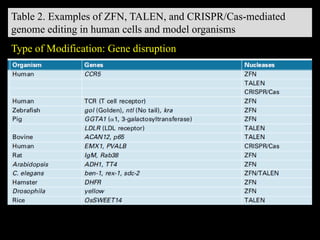
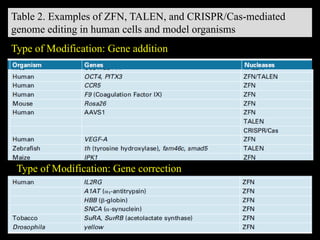
The document discusses various genome editing techniques including meganucleases, zinc fingers, TALENs, and CRISPR-Cas9. It provides details on the mechanism of action, design, advantages, and limitations of each technique. Meganucleases were among the earliest tools but were difficult to engineer for new target sites. Zinc fingers and TALENs improved targeting ability but were still complex to design. CRISPR-Cas9 is now the most widely used system due to its simple and affordable design, high efficiency, and ability to minimize off-target effects.