Downloaded 15 times

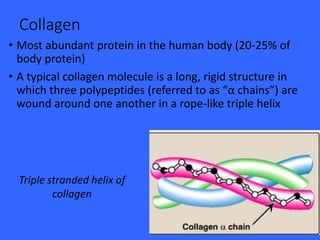




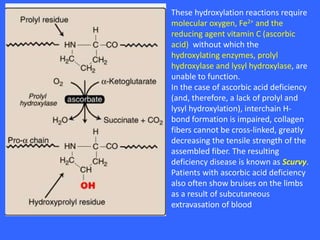


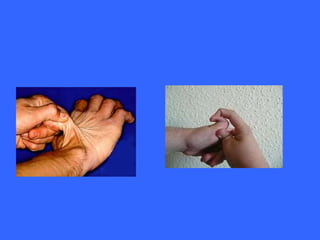



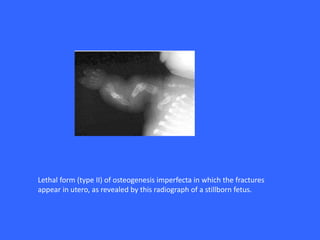


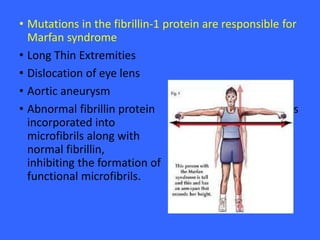







Collagen and elastin are the most abundant fibrous proteins in the body. Collagen forms long, rigid triple helix structures that provide tensile strength. There are over 25 types of collagen that differ in composition but are 1000 amino acids long with repeating Gly-X-Y sequences. Defects in collagen synthesis can cause diseases like Ehlers Danlos syndrome or osteogenesis imperfecta. Elastin provides elasticity and is formed from the protein tropoelastin that aggregates into fibers. Mutations in fibrillin cause Marfan syndrome and deficiencies in alpha-1 antitrypsin can lead to emphysema due to degradation of elastin in the lungs.























































































