Downloaded 15 times
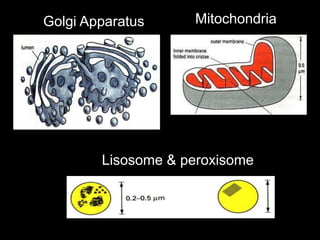

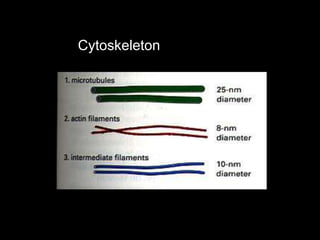
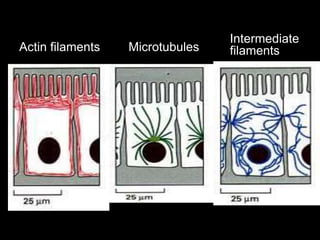

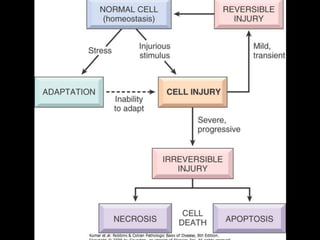
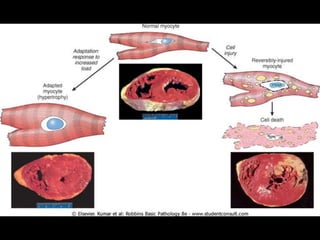


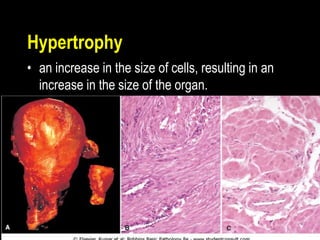

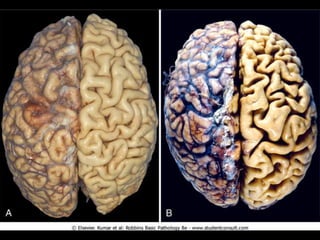
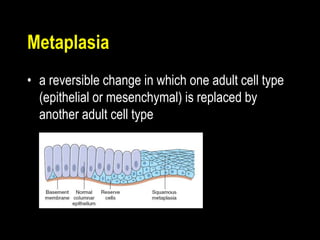
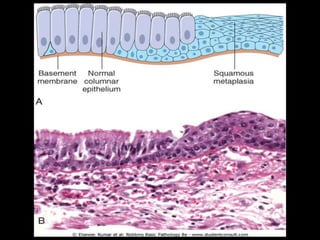


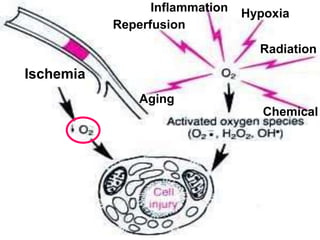

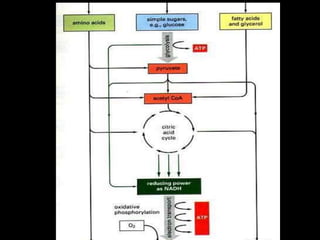

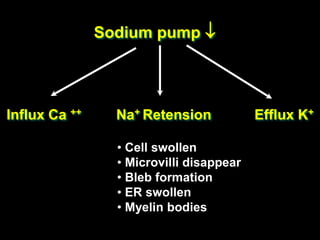

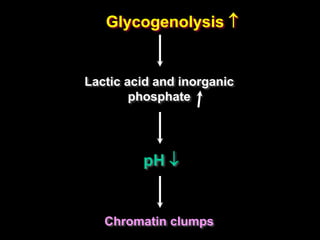

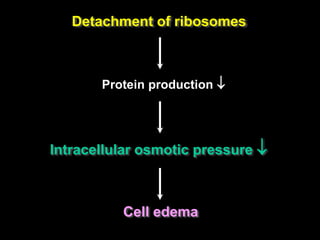
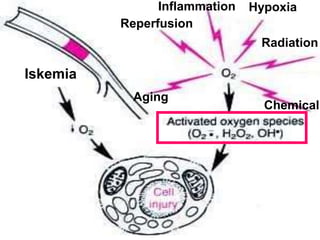



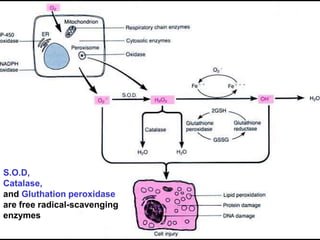


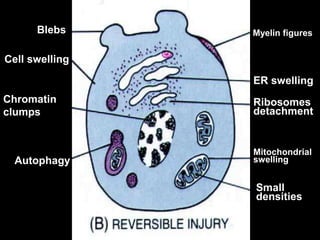
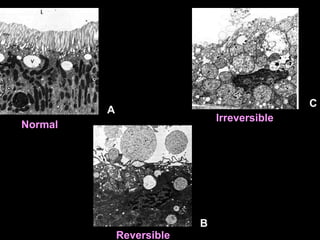

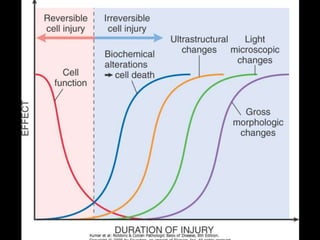
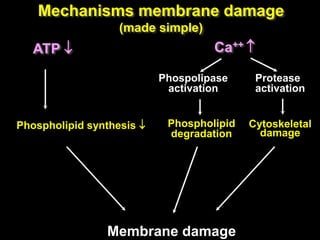
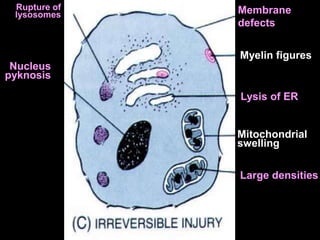


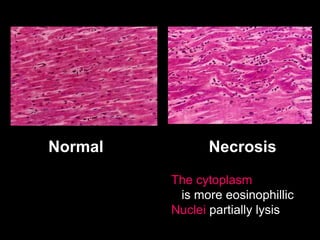
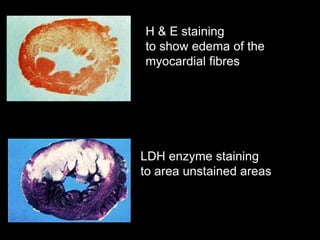
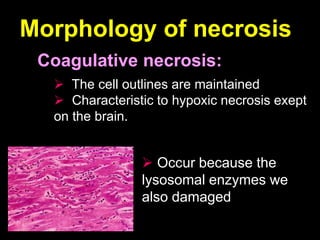
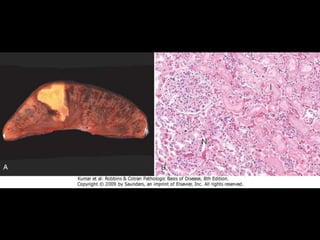

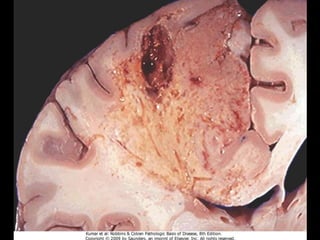
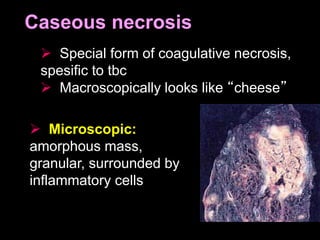

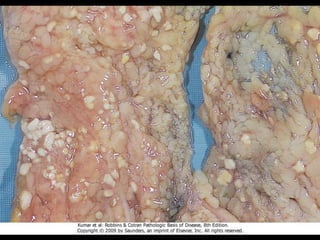
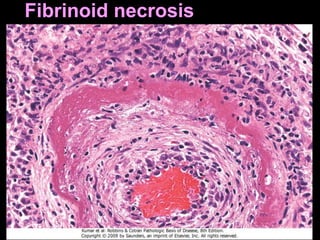

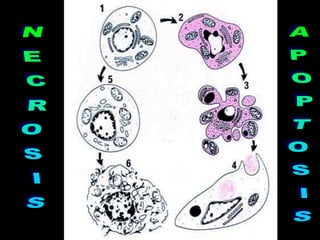
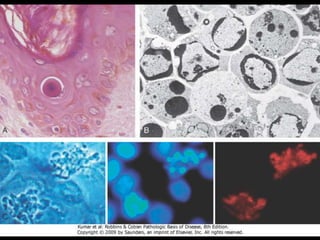
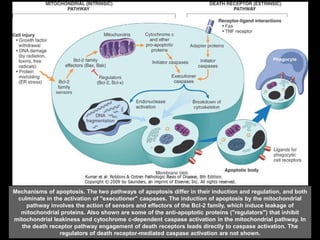
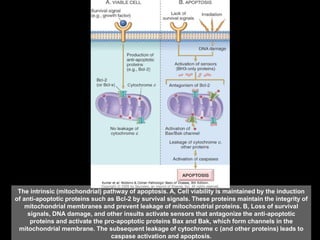
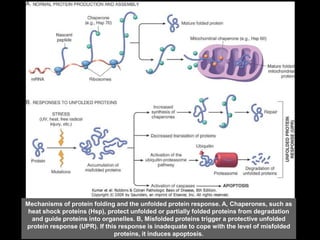
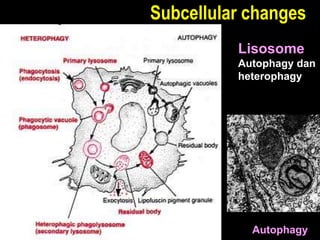
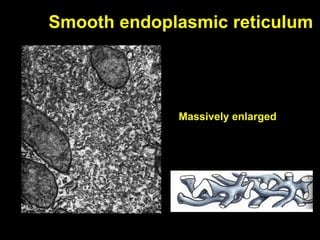
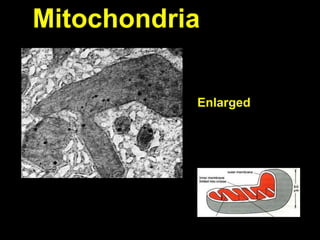
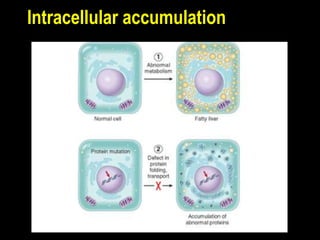
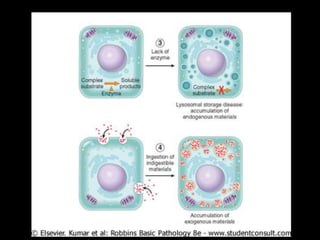
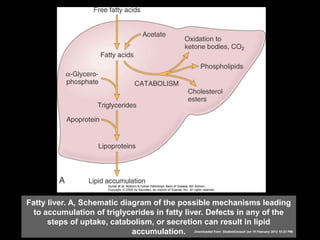
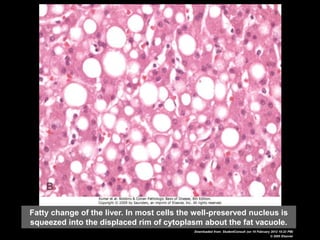
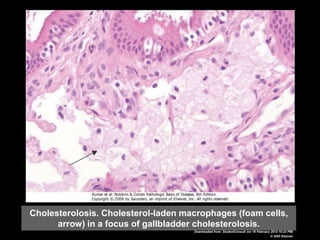
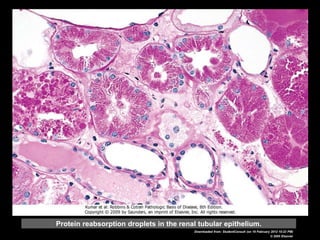
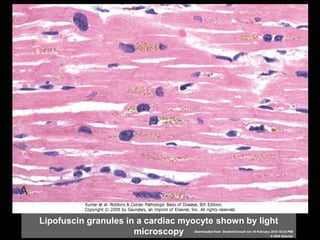
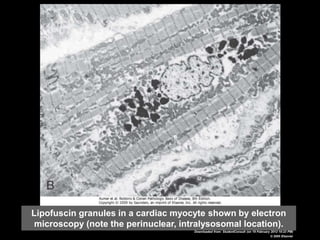
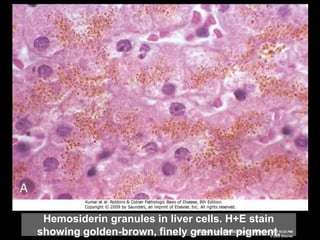
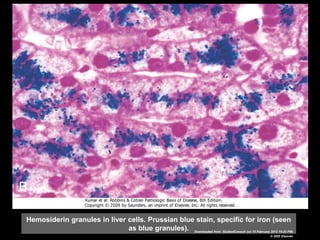




This document discusses cellular adaptation, injury, and death. It covers topics like hyperplasia, hypertrophy, atrophy, metaplasia, causes of cell injury including hypoxia and free radicals, necrosis and apoptosis. It provides detailed descriptions of the morphological changes that occur during cellular injury and the mechanisms of necrosis, apoptosis and intracellular accumulation.




















































































