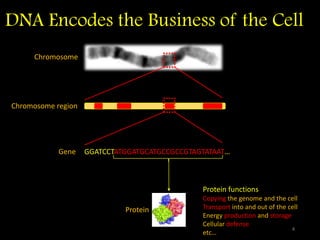
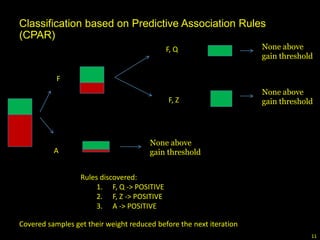
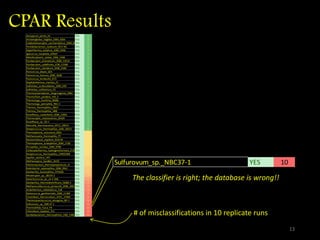
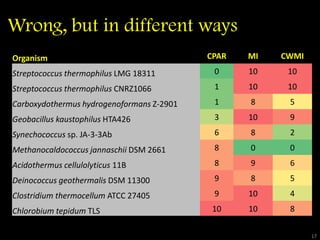
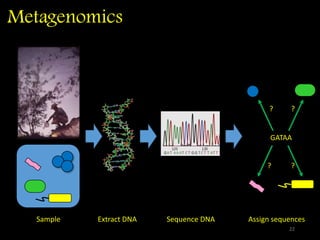
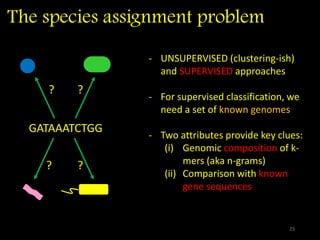
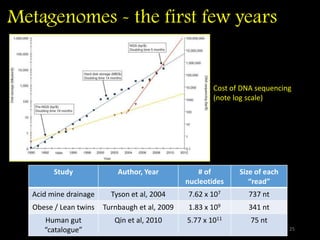
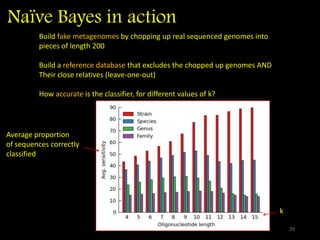
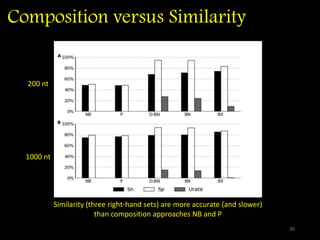
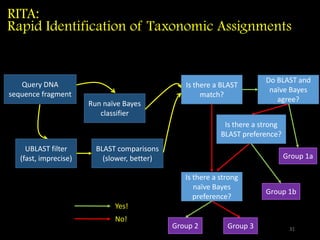
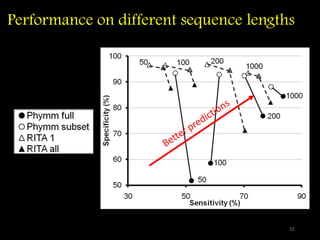
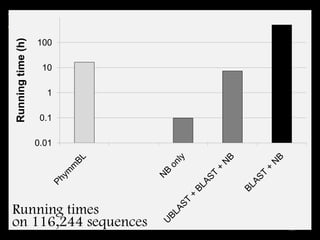
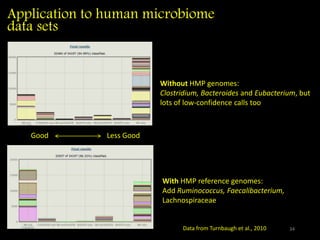
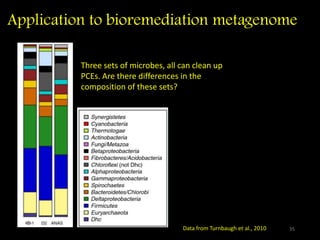
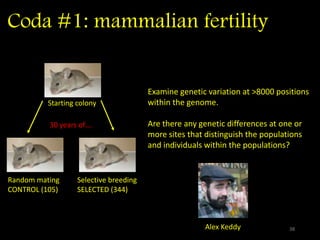
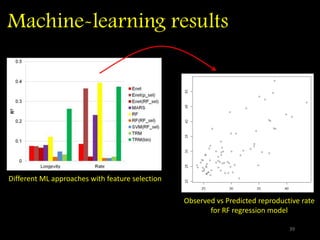
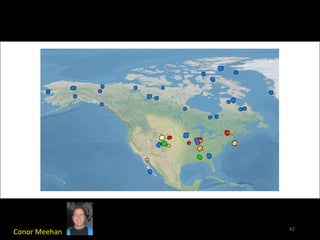
The document discusses classifying biological information from DNA sequences and summarizes some of the challenges involved. It notes that classifying organisms based on their genomes involves determining which genes are responsible for unique properties and can identify organisms. Machine learning techniques like association rule mining are used to discover relationships between genes and phenotypes. However, classifying organisms is challenging due to interactions between genes and effects of taxonomy. Metagenomics, which involves classifying short DNA sequences from environmental samples, poses additional challenges due to analyzing mixtures of many microbial species simultaneously.