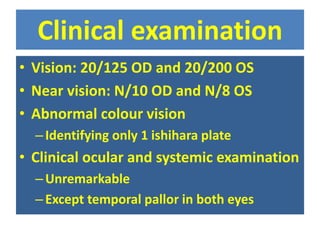
A 30-year-old male presented with progressive bilateral central vision loss since age 10. Examination found vision of 20/125 OD and 20/200 OS with abnormal color vision and temporal pallor in both eyes. His sister had similar symptoms. The initial diagnosis was hereditary optic neuropathy. Differential diagnoses included Leber's hereditary optic neuropathy (LHON), dominant optic atrophy, and other hereditary causes. LHON was thought most likely given the patient's male sex, bilateral symmetric involvement, and positive family history. Further investigation was recommended to confirm the diagnosis.