Downloaded 523 times





























![ differentiate with PD, [123]meta-iodobenzylguanidine
(MIBG) and 123I-iodobenzamide (IBZM) SPECT may be
useful
MIBG is abnormal in PD because of postganglionic
sympathetic denervation, but is typically normal in PSP.
IBZM SPECT assessing the postsynaptic receptors is
abnormal in PSP and normal in PD
IBZM SPECT is abnormal in all APs and therefore
cannot differentiate between PSP and other AP
Novel diagnostic approaches and biomarkers
- csf tau protein
-neurofilament light chain](https://image.slidesharecdn.com/atypicalparkinsonism-150224132309-conversion-gate02/85/Atypical-parkinsonism-34-320.jpg)


















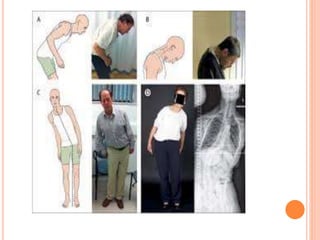
![ Orofacial dystonia or dyskinesias
Atypical spontaneous or levodopa induced
dystonia
dyskinesia mainly affecting orofacial muscles,
[resembling risus sardonicus of cephalic tetanus]
Axial dystonia -Pisa syndrome (subacute axial
dystonia with a severe tonic lateral flexion of the
trunk, head, and neck)
early severe camptocormia
Disproportionate antecollis -Chin on chest, neck
can only be passively and forcibly extended to its
normal position with difficulty; despite severe
chronic neck flexion, flexion elsewhere is minor.](https://image.slidesharecdn.com/atypicalparkinsonism-150224132309-conversion-gate02/85/Atypical-parkinsonism-56-320.jpg)




























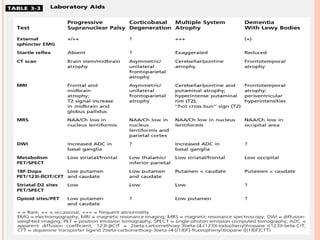
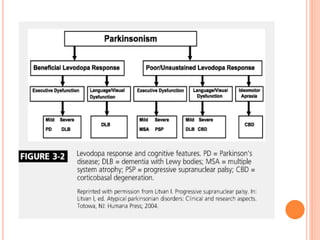
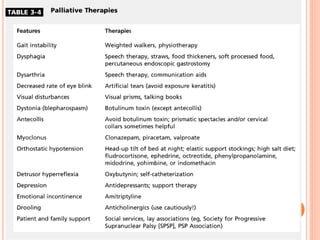

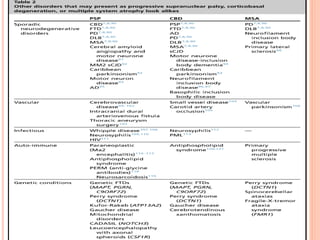



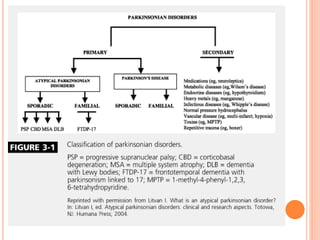
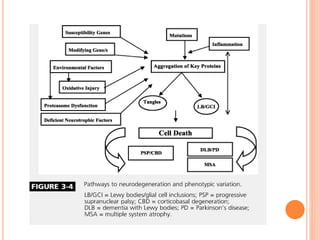
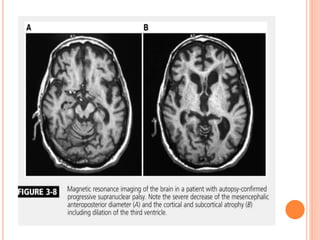
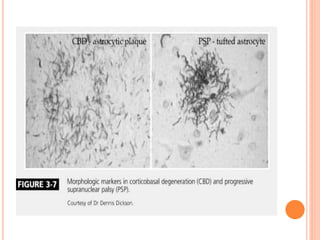
This document discusses atypical parkinsonism (AP), which accounts for 15-20% of all cases of parkinsonism. It begins by classifying AP into primary, multisystem degenerations, hereditodegenerative, and secondary types. It then focuses on the major AP syndromes - Progressive Supranuclear Palsy (PSP), Multiple System Atrophy (MSA), and Corticobasal Degeneration (CBD). For PSP and MSA, it provides details on clinical presentation, diagnostic criteria, investigations, pathology, and treatment approaches. The document emphasizes the importance of differentiating AP from Parkinson's disease to allow for accurate prognosis and management of patients.































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)







