Downloaded 49 times



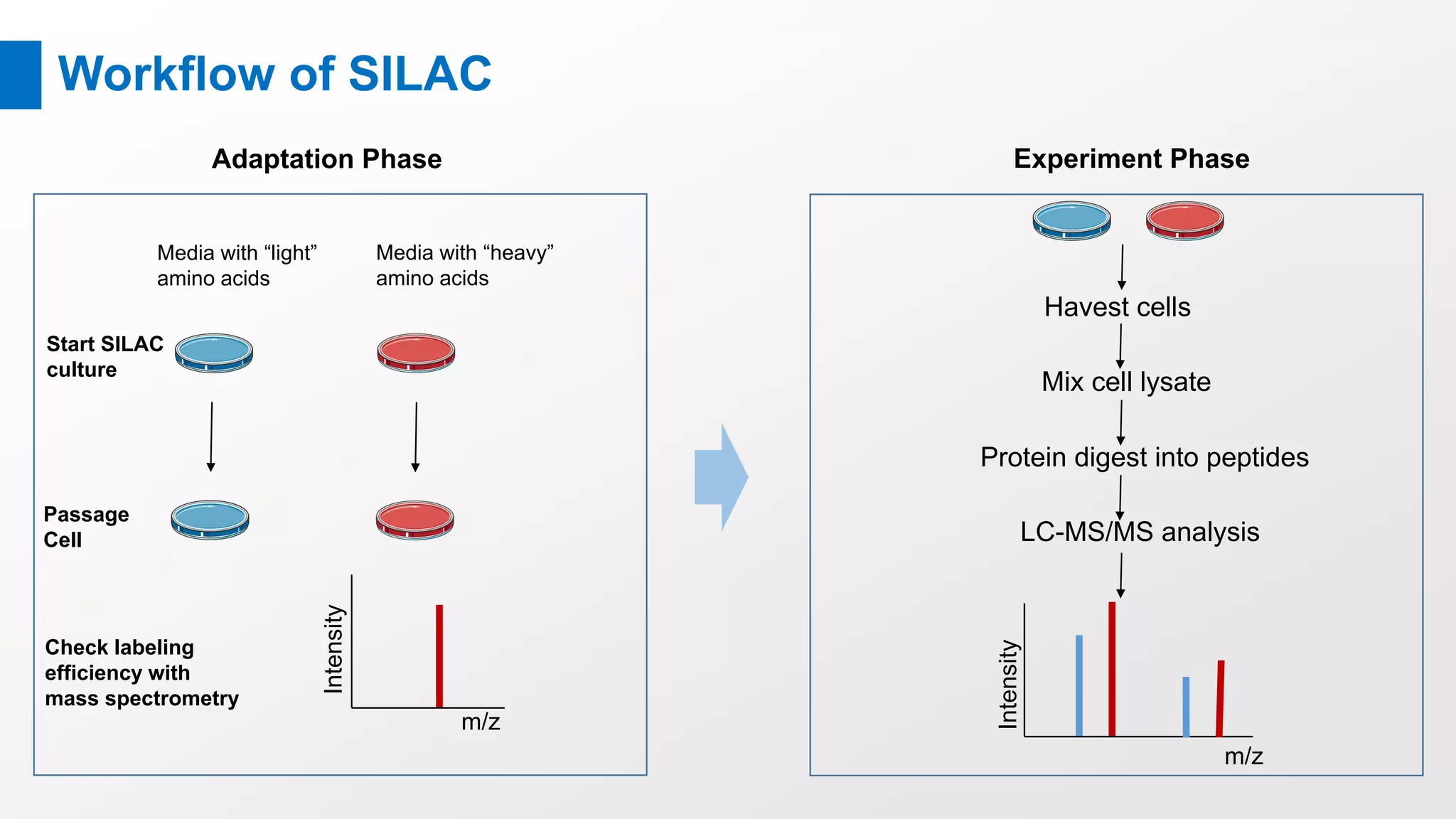
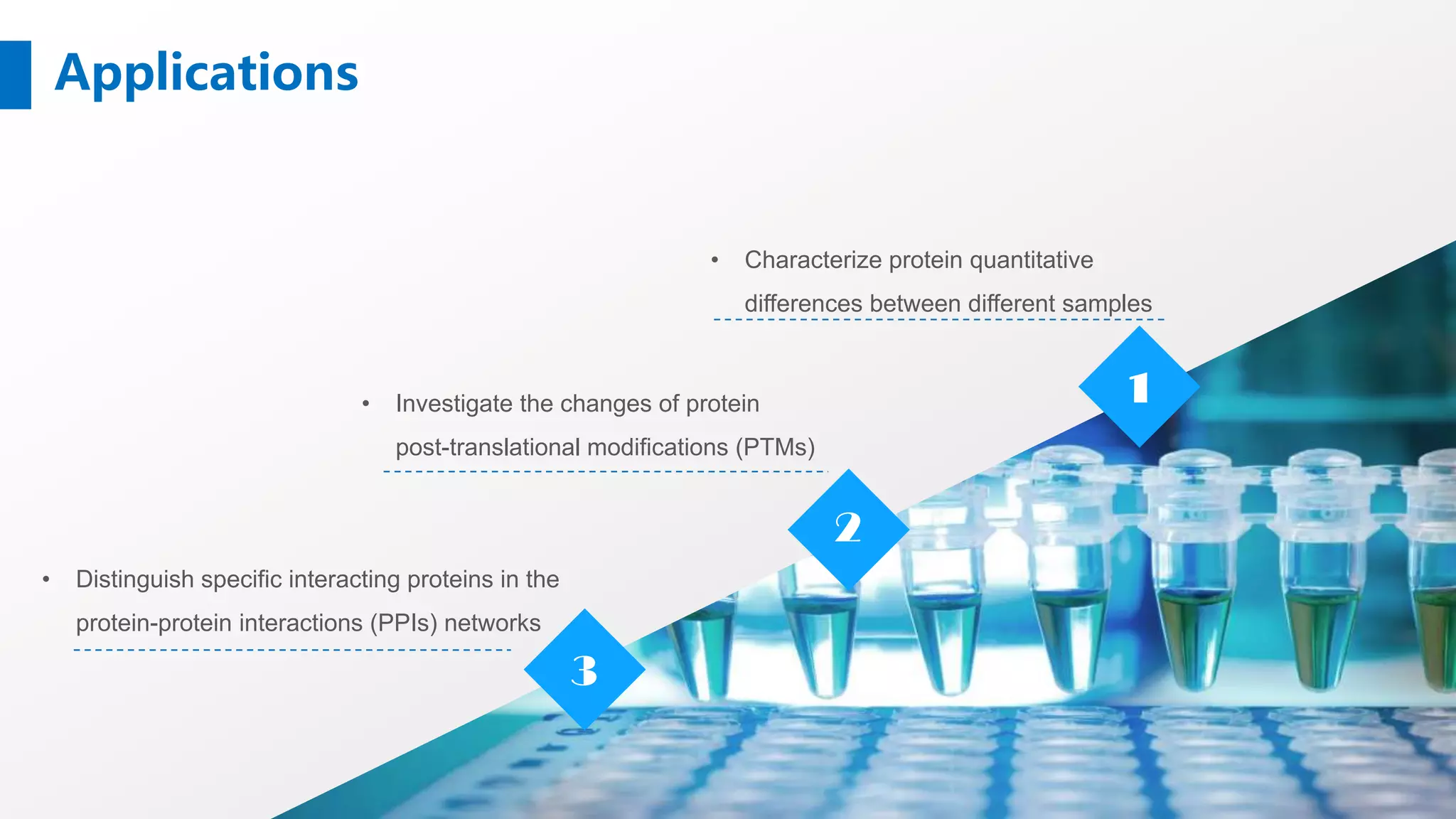
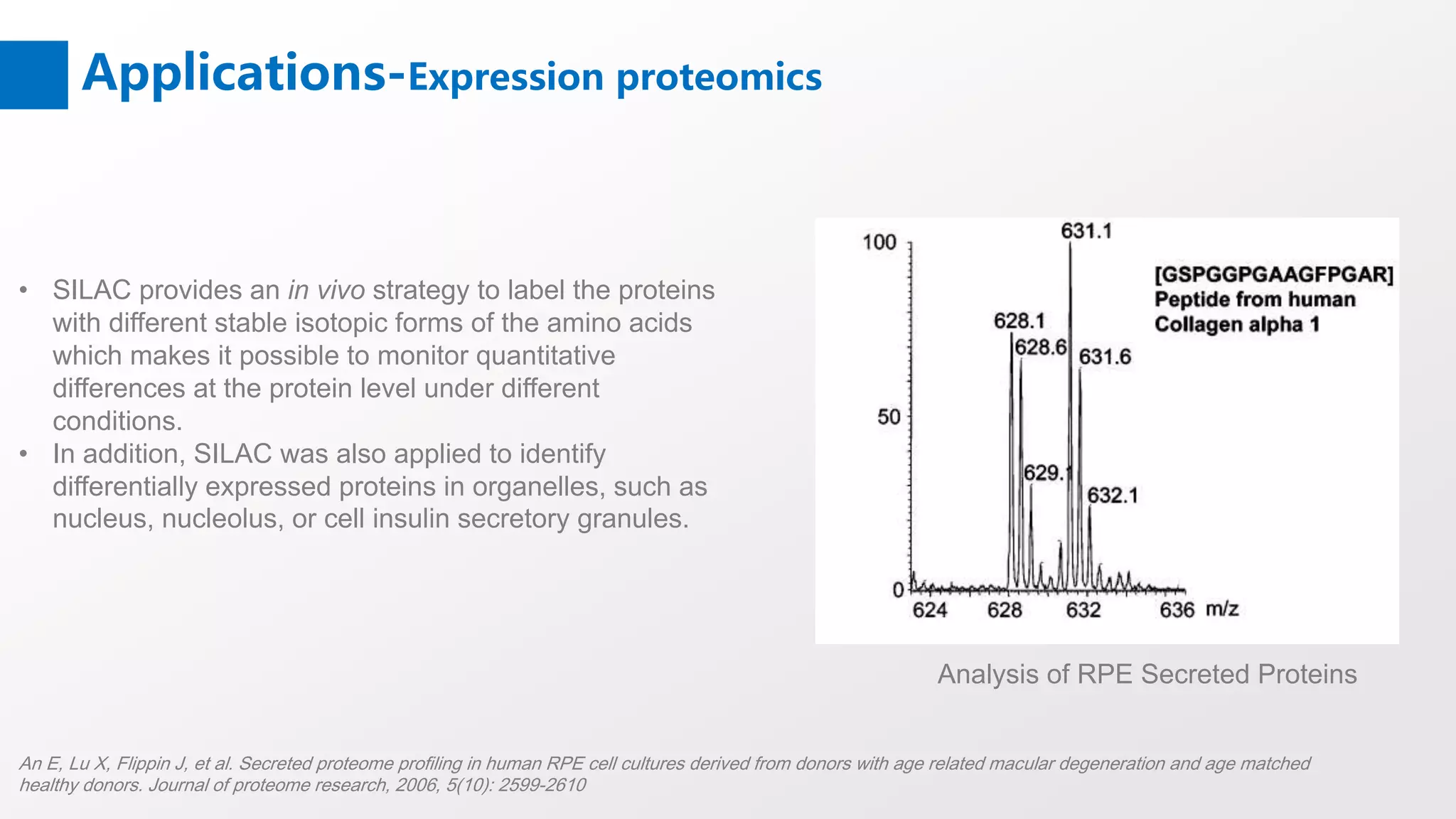

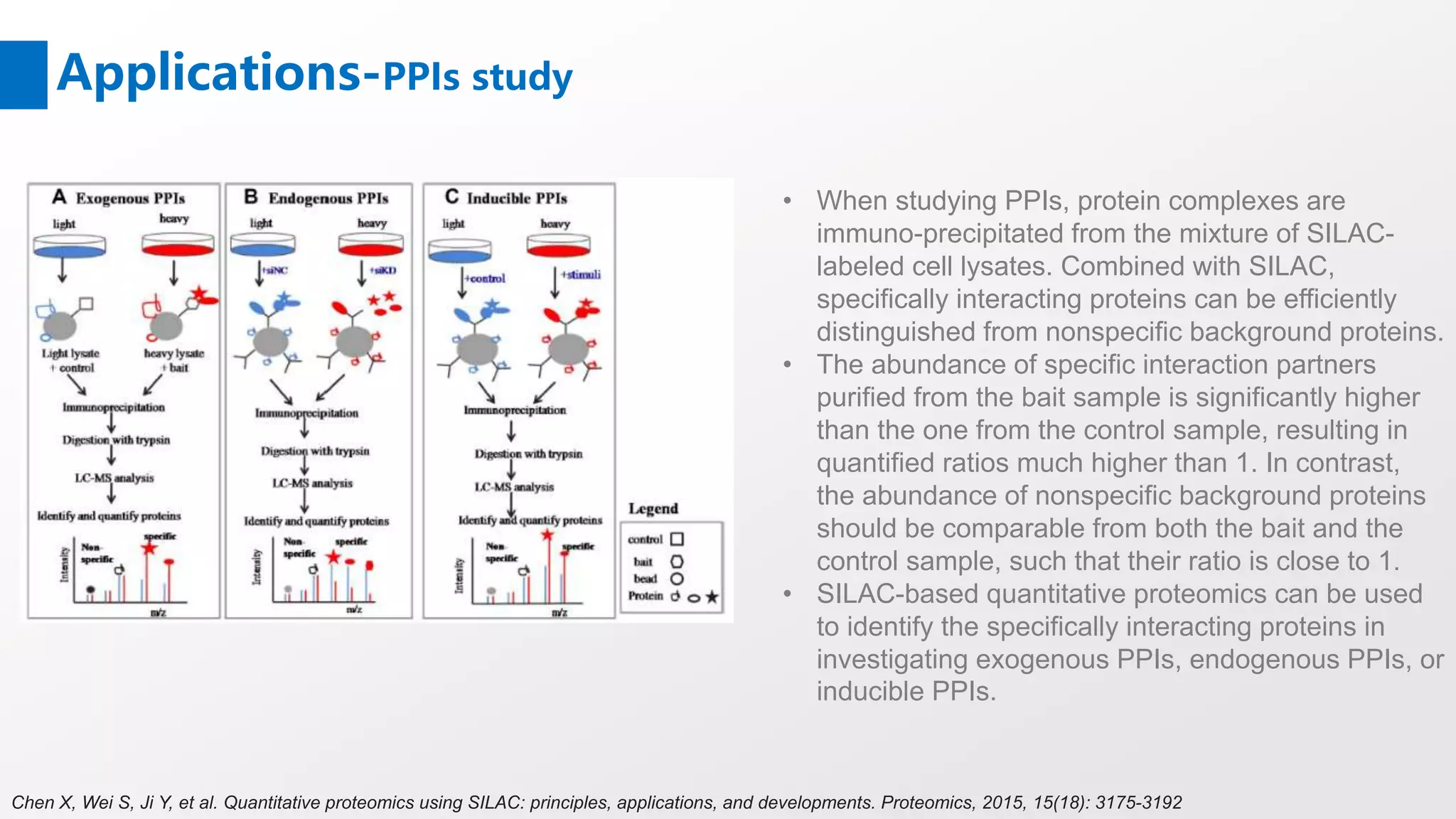




Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) is a mass spectrometry technique that enables accurate quantification of protein abundance changes without chemical manipulation. It involves incorporating 'light' and 'heavy' labeled amino acids into proteins, allowing for comparative analysis of different samples, post-translational modifications, and protein-protein interactions. Creative Proteomics offers various SILAC and proteomics quantification services to enhance research accuracy and reliability.






















































































