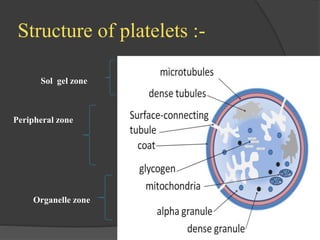
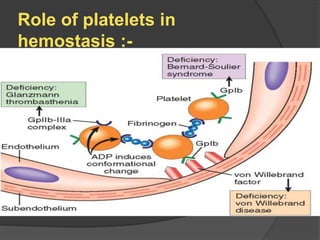
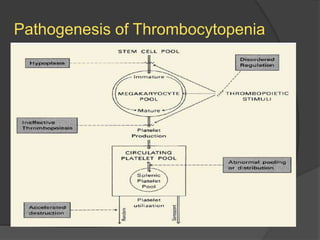
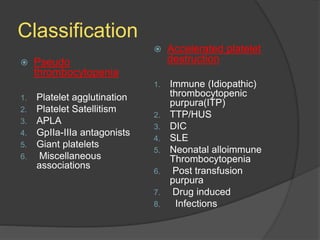
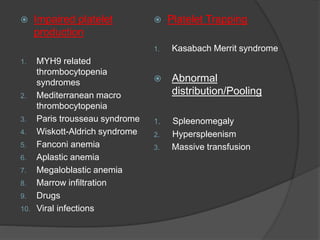
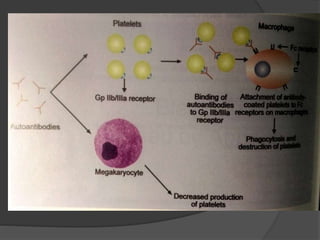
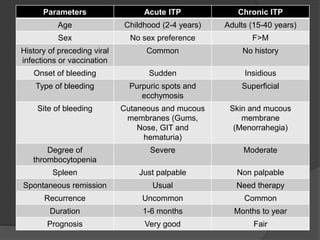
Platelets were first observed in 1841 and were termed "platelets" in 1882. Their role in hemostasis through adhesion, secretion, and aggregation was established over the late 19th century. Megakaryocytes were recognized as rare bone marrow cells that produce platelets, with this relationship established in 1906. Thrombocytopenia can result from impaired platelet production, increased platelet destruction, or platelet sequestration. It is classified based on etiology, with immune thrombocytopenia and drug-induced thrombocytopenia as common causes of accelerated platelet destruction.