This presentation involves bio equivalence studies and how it is designed . Bioequivalence refers to the absence of a significant difference in the rate and extent of drug absorption between two pharmaceutical products when administered at the same molar dose under similar conditions.
Two drug products are considered bioequivalent if their pharmacokinetic (PK) parameters, such as:
Maximum plasma concentration (Cmax)
Area under the plasma concentration-time curve (AUC)
Time to reach Cmax (Tmax)
Bioequivalence Studies: Overview, Methods, and Clinical Significance
Bioequivalence (BE) studies are conducted to demonstrate that a generic drug is therapeutically equivalent to its brand-name (reference) drug. These studies compare the pharmacokinetics (PK) of both drugs to ensure they provide the same efficacy and safety when administered at the same dosage under similar conditions.
---
1. Objectives of Bioequivalence Studies
To confirm that the generic drug delivers the same rate and extent of absorption as the reference drug.
To ensure interchangeability between generic and brand-name drugs without affecting therapeutic outcomes.
To avoid extensive clinical trials while maintaining drug safety and efficacy.
---
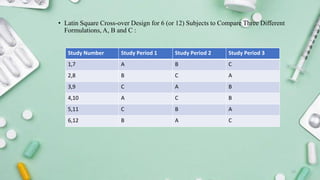
2. Study Design of Bioequivalence Trials
a. Study Type
Single-dose, two-treatment, two-period, two-sequence crossover study (most common).
Parallel design (used for long half-life drugs).
Replicated crossover design (used for drugs with high variability).
b. Study Population
Healthy volunteers (typically 18-55 years).
Sometimes special populations (e.g., elderly, renal/hepatic impairment) are considered.
c. Dosing and Administration
Single-dose administration of both test (generic) and reference (brand) formulations.
Washout period between doses to eliminate carryover effects.
d. Pharmacokinetic (PK) Parameters Measured
Maximum plasma concentration (Cmax): Measures drug absorption rate.
Area under the curve (AUC0-t, AUC0-∞): Represents total drug exposure.
Time to reach Cmax (Tmax): Indicates the speed of drug absorption.
Elimination half-life (t½): Evaluates how long the drug stays in the system.
e. Acceptance Criteria
Regulatory agencies (e.g., FDA, EMA, WHO) require that the 90% confidence interval (CI) for the ratio of the test to reference drug falls within 80%-125% for Cmax and AUC.
Stricter criteria (90%-111%) apply for narrow therapeutic index (NTI) drugs (e.g., warfarin, digoxin).
---
3. Methods Used in Bioequivalence Studies
a. Pharmacokinetic Studies (In Vivo Bioequivalence)
Blood/plasma samples are collected at various time points post-dosing.
Liquid Chromatography-Mass Spectrometry (LC-MS) is used for drug concentration analysis.
b. In Vitro Dissolution Testing
Assesses how the drug dissolves under physiological conditions.
Ensures that the drug releases its active ingredient similarly to the reference drug.
c. Biowaivers (In Vitro Bioequivalence)




















![REFERENCES
• Brahmankar HA, Jaiswal SB. Biopharmaceutics and pharmacokinetics: a treatise.
2nd ed. New Delhi: Vallabh Prakashan; 2009.
• European Medicines Agency. Guideline on the investigation of bioequivalence
[Internet]. London: EMA; 2010
21](https://image.slidesharecdn.com/bioequivalence-250404173559-c5dcd844/85/BIOEQUIVALENCE-studies-In-biopharmaceutics-21-320.jpg)
















































