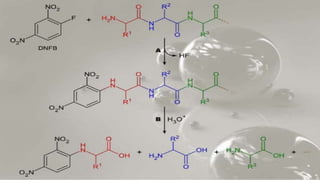
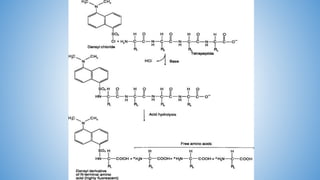
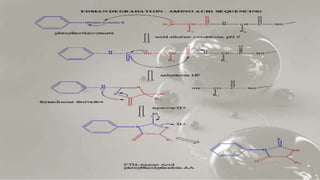
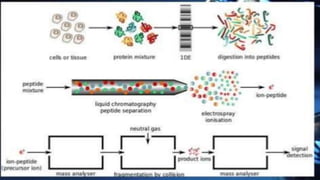
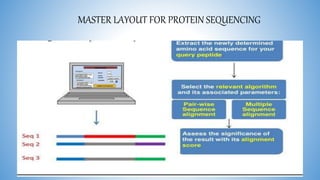
The document discusses various methods for protein sequence analysis, including: (1) N-terminal sequencing using Edman degradation, (2) C-terminal sequencing using carboxypeptidases, (3) DNA sequencing to infer protein sequence, and (4) mass spectrometry. It also covers preparing proteins for sequencing by separating chains and cleaving disulfide bridges, as well as bioinformatics tools like BLAST for comparing sequences. The overall goal of protein sequencing is to determine amino acid sequences to understand protein structure, function, and cellular processes.