Downloaded 756 times
















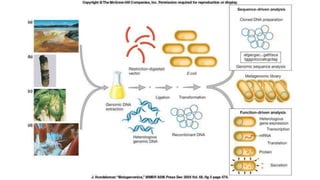




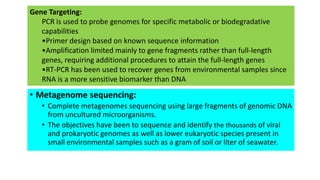
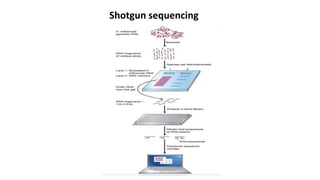

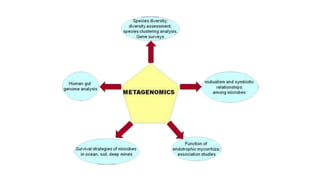





This document discusses the field of metagenomics, which involves directly extracting and sequencing genetic material from environmental samples without culturing individual microbial species. It provides a brief history of metagenomics from early microbiologists in the 17th century to recent large-scale sequencing projects. Methods of metagenomic analysis like sequence-driven and function-driven approaches are described. Applications to studying uncultured symbiotic microbes, extreme environments, and the human gut microbiome are also summarized.














































![[2013.12.02] Mads Albertsen: Extracting Genomes from Metagenomes](https://cdn.slidesharecdn.com/ss_thumbnails/2013-131202013655-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)














![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)
















