Презентація вебінару "IVDR. Що це за звір та коли буде в Україні?", 29.07.2022
- Регулювання 2017/746 для медичних виробів in vitro, основні зміни;
- Зміна класифікації виробів, окрема класифікація для виробів аналізу на COVID: чому?
- Чи варто очікувати оновлення регулювання в Україні?
ISO 13485:2016 andIVDR
Новий IVDR слід розглядати разом з EN ISO
13485:2016 адже включає одразу декілька
перехресних посилань (наприклад, додатки до
IVDD!)
• 4.2.3 Технічний файл медичного виробу
• 5.4 Цілі якості стосуються зобов'язань, які
необхідно виконати
УСІ відповідні нормативні вимоги (REACH, WEE,
RoHs, CLP, GDPR)
• Необхідно створити План якості або еквівалент
6.
Quality planning
Чи єу вас план щодо якості
для документування переходу
від IVDD до IVDR? Чи
проводили ви аналіз СУЯ,
щоб визначити потенційні
прогалини між IVDD та IVDR?
Як діяти у випадку
невведнення еквіваленту
IVDR на території України,
але зобов’язання ввести на
території ЄС?
7.
Поняття in vitromedical device
• ‘in vitro diagnostic medical device’ means any medical
device which is a reagent, reagent product, calibrator,
control material, kit, instrument, apparatus, equipment, or
system, whether used alone or in combination, intended by
the manufacturer to be used in vitro for the examination of
specimens, including blood and tissue donations, derived
from the human body, solely or principally for the purpose
of providing information:
• concerning a physiological or pathological state,
• concerning a congenital abnormality, or
• to determine the safety and compatibility with potential
recipients, or
• to monitor therapeutic measures.
‘in vitro diagnostic medical device’ means any medical
device which is a reagent, reagent product, calibrator,
control material, kit, instrument, apparatus, piece of
equipment, software or system, whether used alone or in
combination, intended by the manufacturer to be used in
vitro for the examination of specimens, including blood and
tissue donations, derived from the human body, solely or
principally for the purpose of providing information on one
or more of the following:
(a) concerning a physiological or pathological process or
state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a
disease;
(d) to determine the safety and compatibility with potential
recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
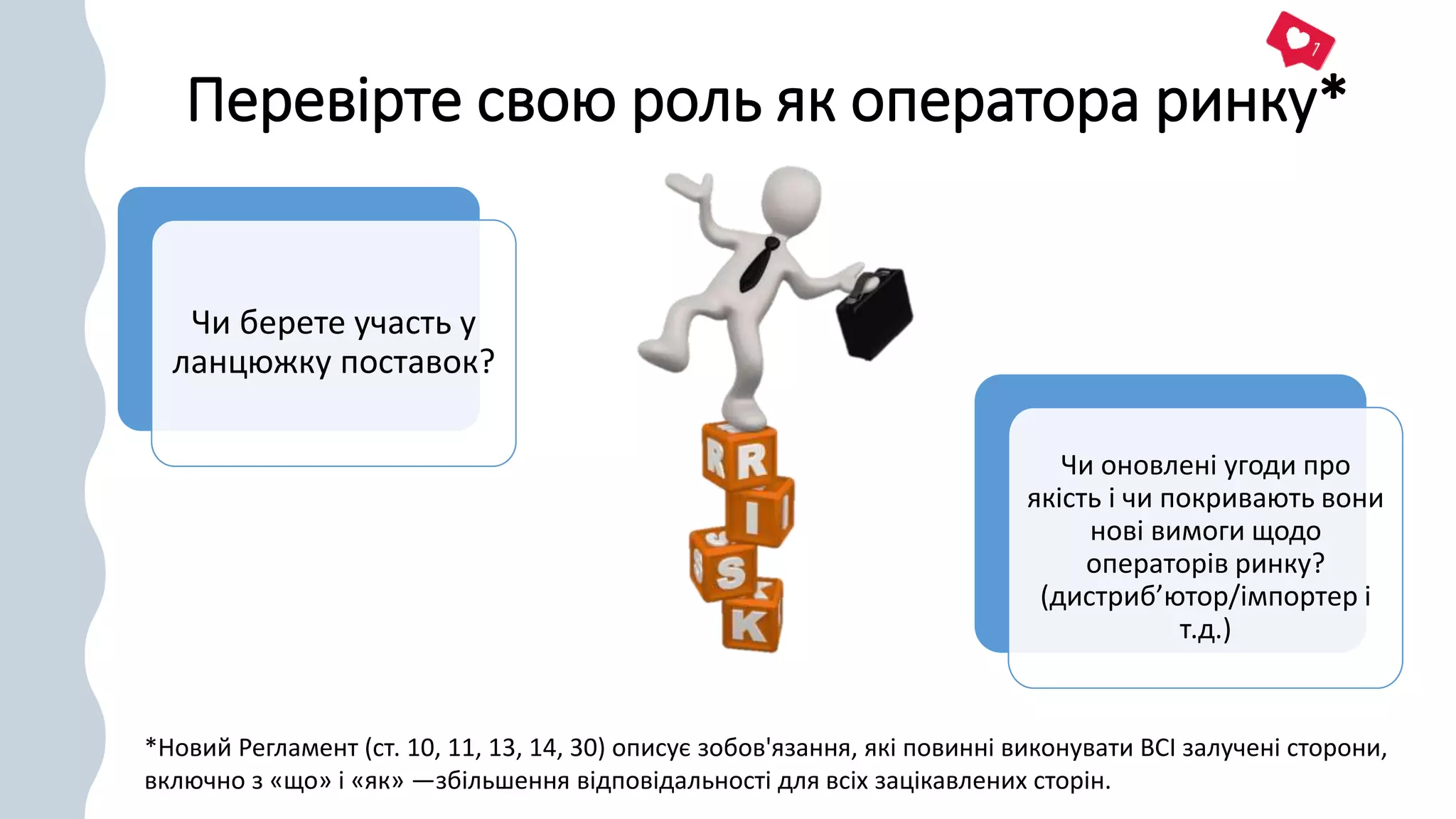
Перевірте свою рольяк оператора ринку*
Чи берете участь у
ланцюжку поставок?
Чи оновлені угоди про
якість і чи покривають вони
нові вимоги щодо
операторів ринку?
(дистриб’ютор/імпортер і
т.д.)
*Новий Регламент (ст. 10, 11, 13, 14, 30) описує зобов'язання, які повинні виконувати ВСІ залучені сторони,
включно з «що» і «як» —збільшення відповідальності для всіх зацікавлених сторін.
Перевірте свій статус
наукової,аналітичної
інформації та даніщодо
клінічної ефективності
Чи є у вас план оцінки
ефективності, розглядаючи
наукову обґрунтованість,
аналітичні та оцінку
клінічної ефективності?
Чи проводяться у вас
клінічні дослідження? Чи
розглядали Ви останні
версії стандартів?
ISO 20916 In vitro diagnostic
medical devices — Clinical
performance studies using
specimens from human
subjects — Good study
practice
та
ISO 14971 Medical devices
— Application of risk
management to medical
devices
Клінічна ефективність?
15.
Клінічна
ефективність
Очікування щодо включенняклінічних даних для IVD зросли
✓ Клінічні заяви повинні бути підтверджені даними та
конкретними звітами залежно від ризику вашого виробу
✓ Клінічні докази мають бути обґрунтовані даними та висновками
✓ Клінічні докази необхідно постійно оновлювати для
забезпечення, що вироби залишаються ефективними та
безпечними. Це включає PMS.
✓ Слідуйте плану оцінки ефективності
✓ Потрібен звіт про оцінку ефективності
✓ Summary of Safety and Performance необхідні для класу C + D
✓ Нотифікований орган розглядатиме не тільки дані, а й
методологію
16.
Перевірте свій
PMS іPMPF
Чи є у вас план
постмаркетингового нагляду?
• Що підтверджує встановлення
проактивного та
систематичного процесу
збирання будь-якої інформації,
пов’язаної з PMS?
• Чи є у вас план подальших
заходів після виходу на ринок,
який?
17.
PMS і PMPF
✓Необхіднідля кожного виробу
✓Підхід на основі життєвого циклу продукту та
клінічних доказів
✓Збір інформації (інциденти, тенденції, скарги,
відгуки тощо) і оціниювання безпечності та
ефективності
✓Систематична оцінка інформації та постійне
управління ризиками
✓Провести клінічну оцінку ризику/користі
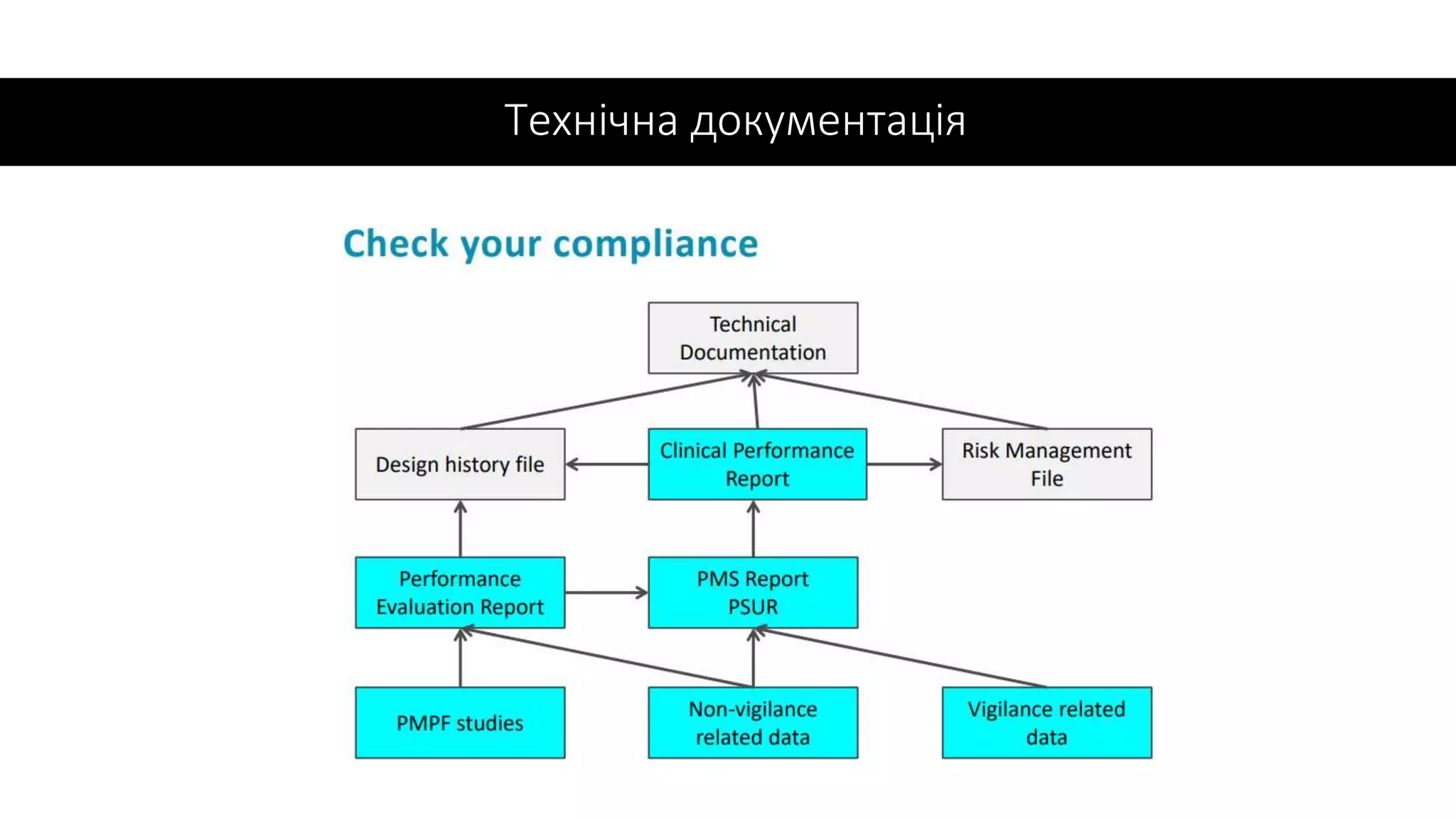
✓Звіт PMS, частина Технічної документації
✓PMPF є частиною PMS як безперервний процес
оновлення даних щодо клінічної ефективності