Downloaded 2,105 times
![Phylogenetic Analysis Caro-Beth Stewart, Ph.D . Associate Professor Department of Biological Sciences University at Albany, SUNY Albany, New York 12222 [email_address] Lecture presented at the National Human Genome Research Institute for the course Current Topics in Genome Analysis 2000](https://image.slidesharecdn.com/phylogeneticanalysis-111117220939-phpapp01/75/Phylogenetic-analysis-1-2048.jpg)




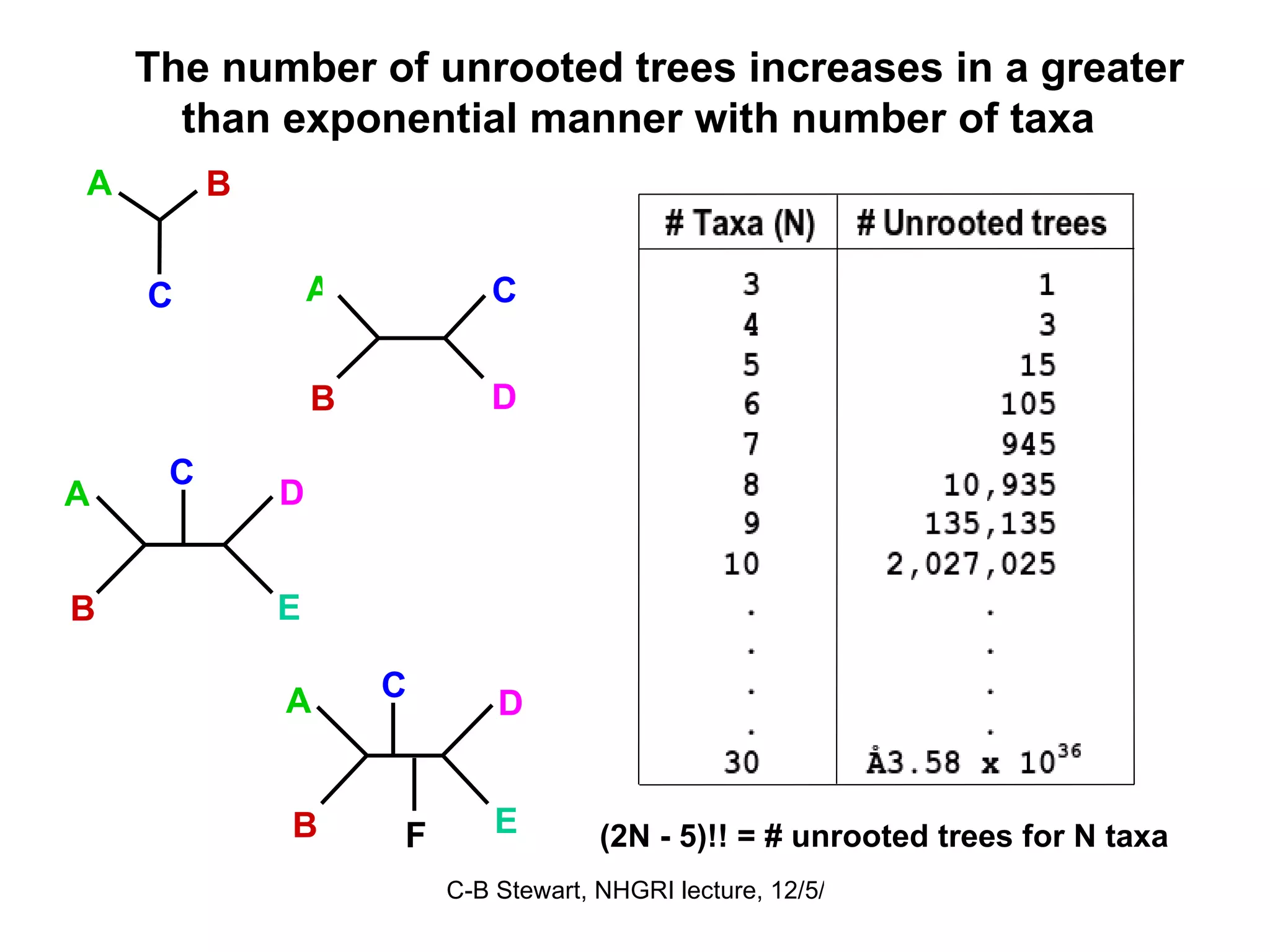
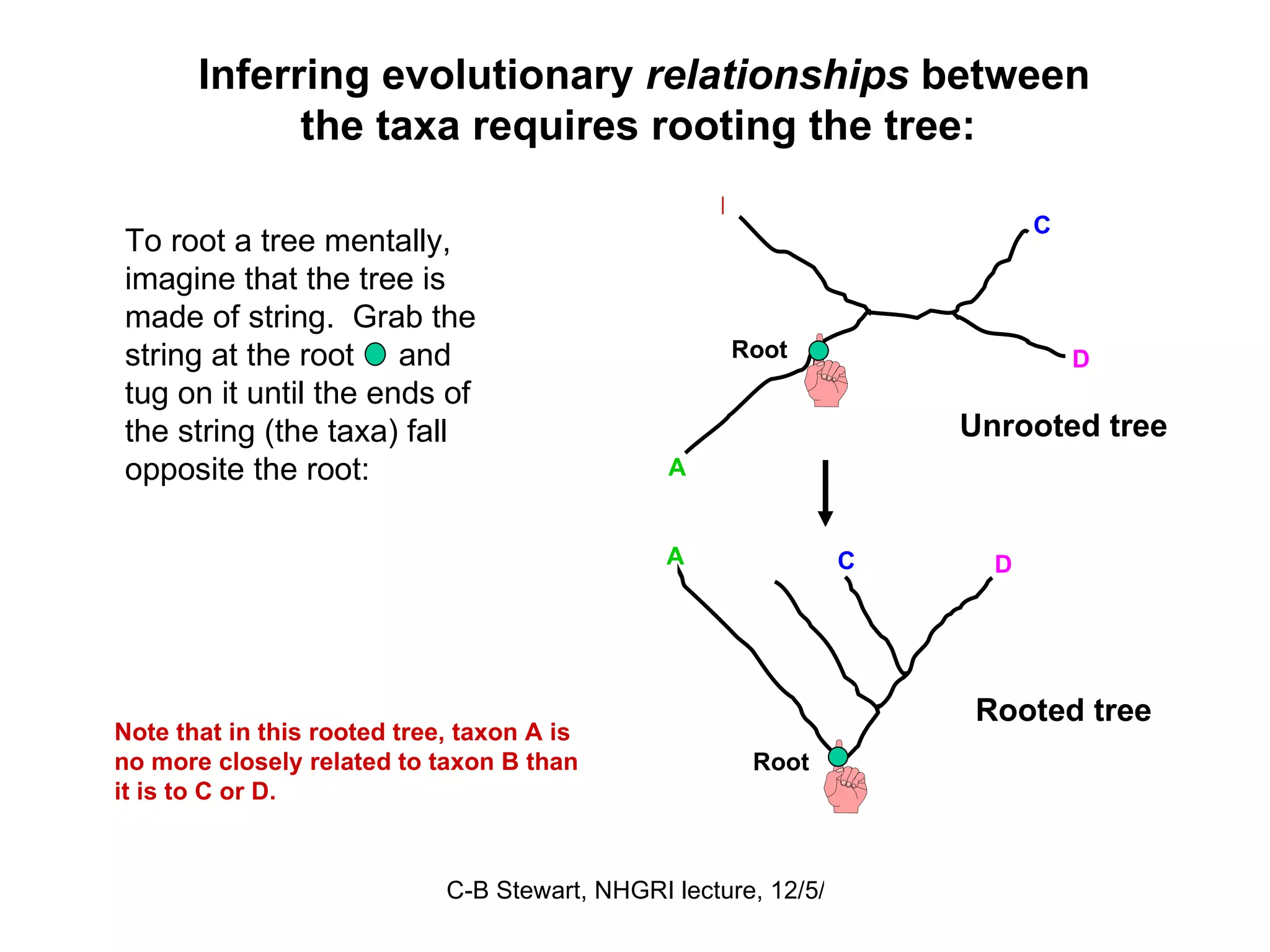
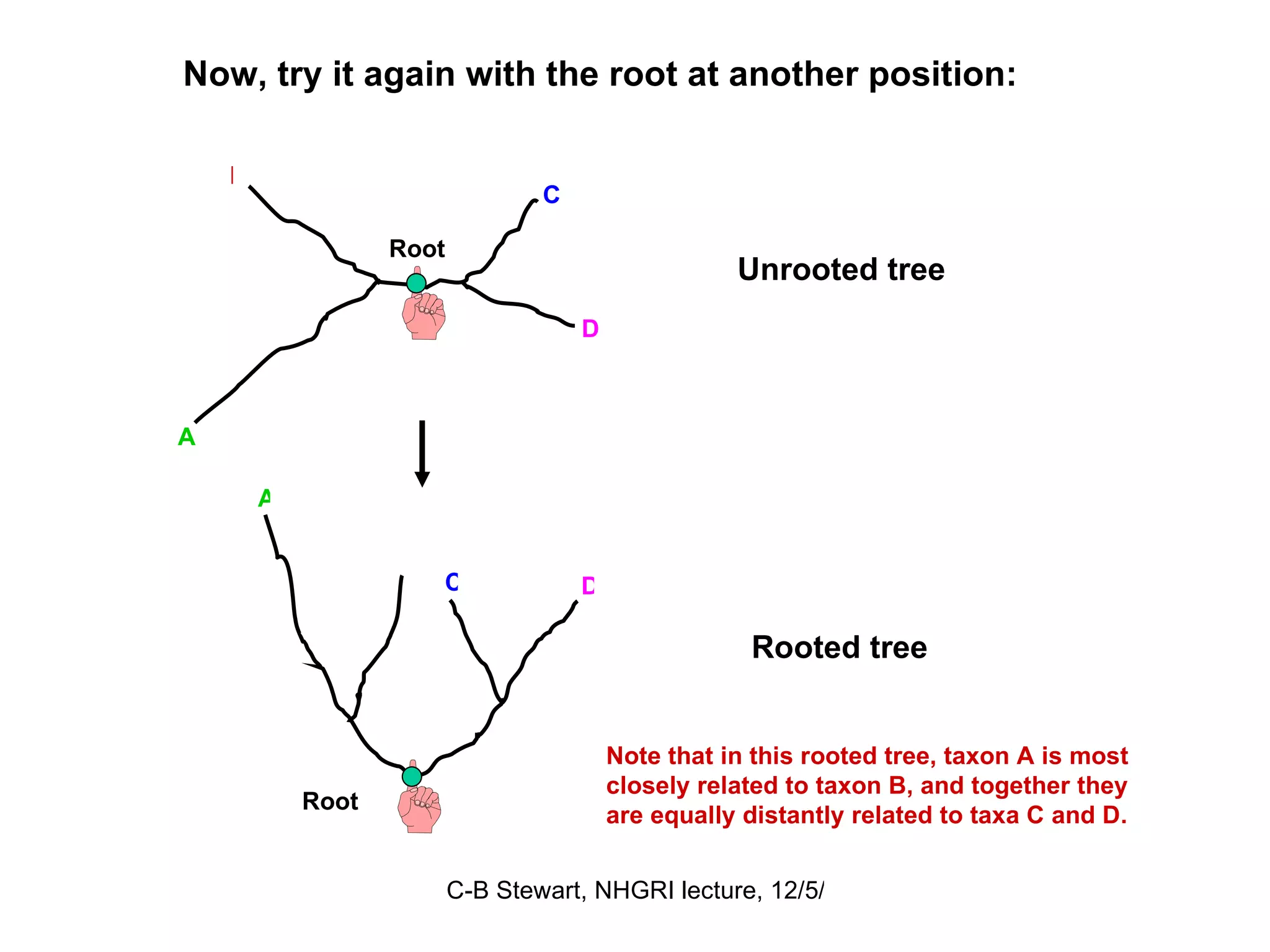
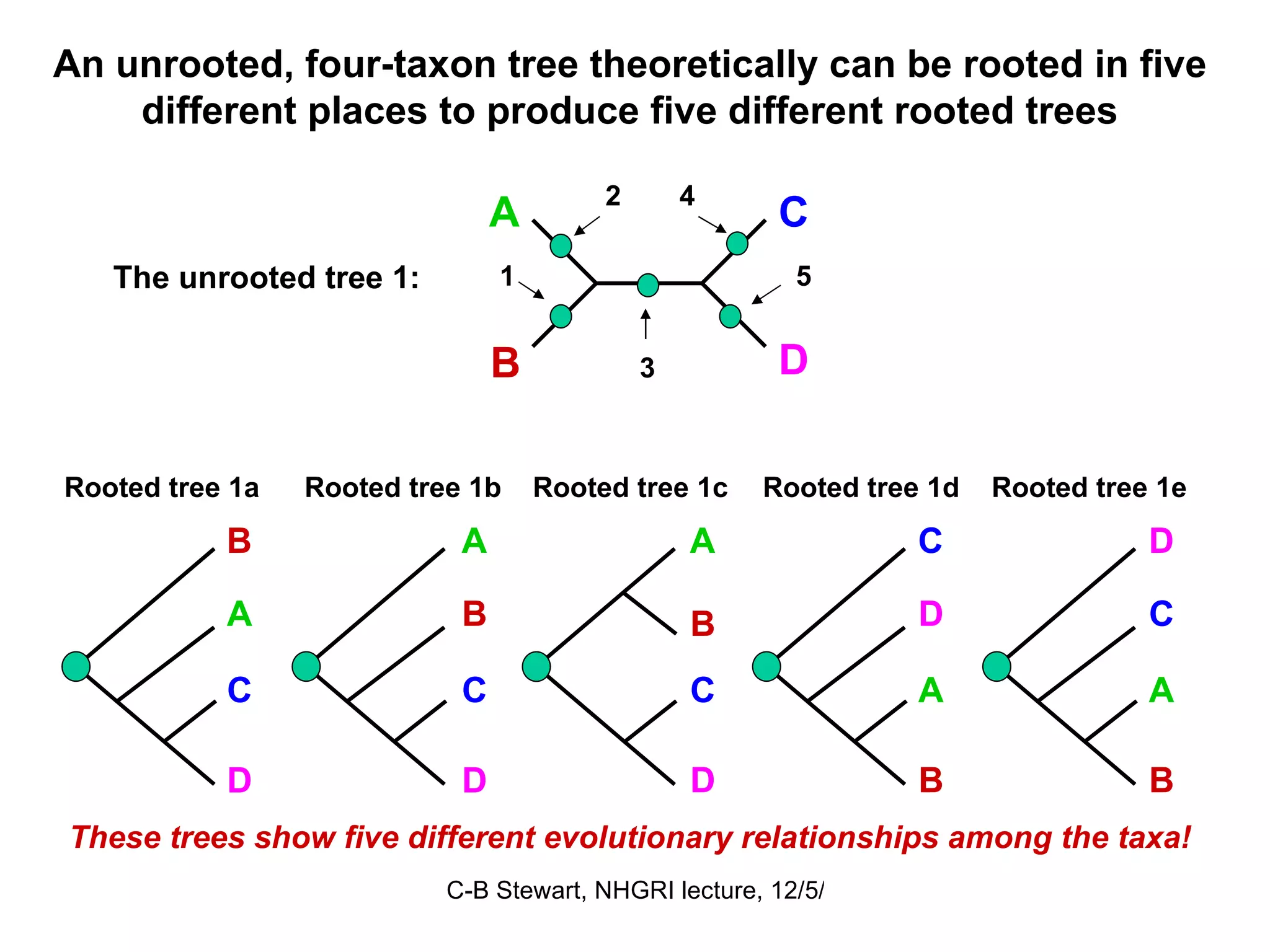

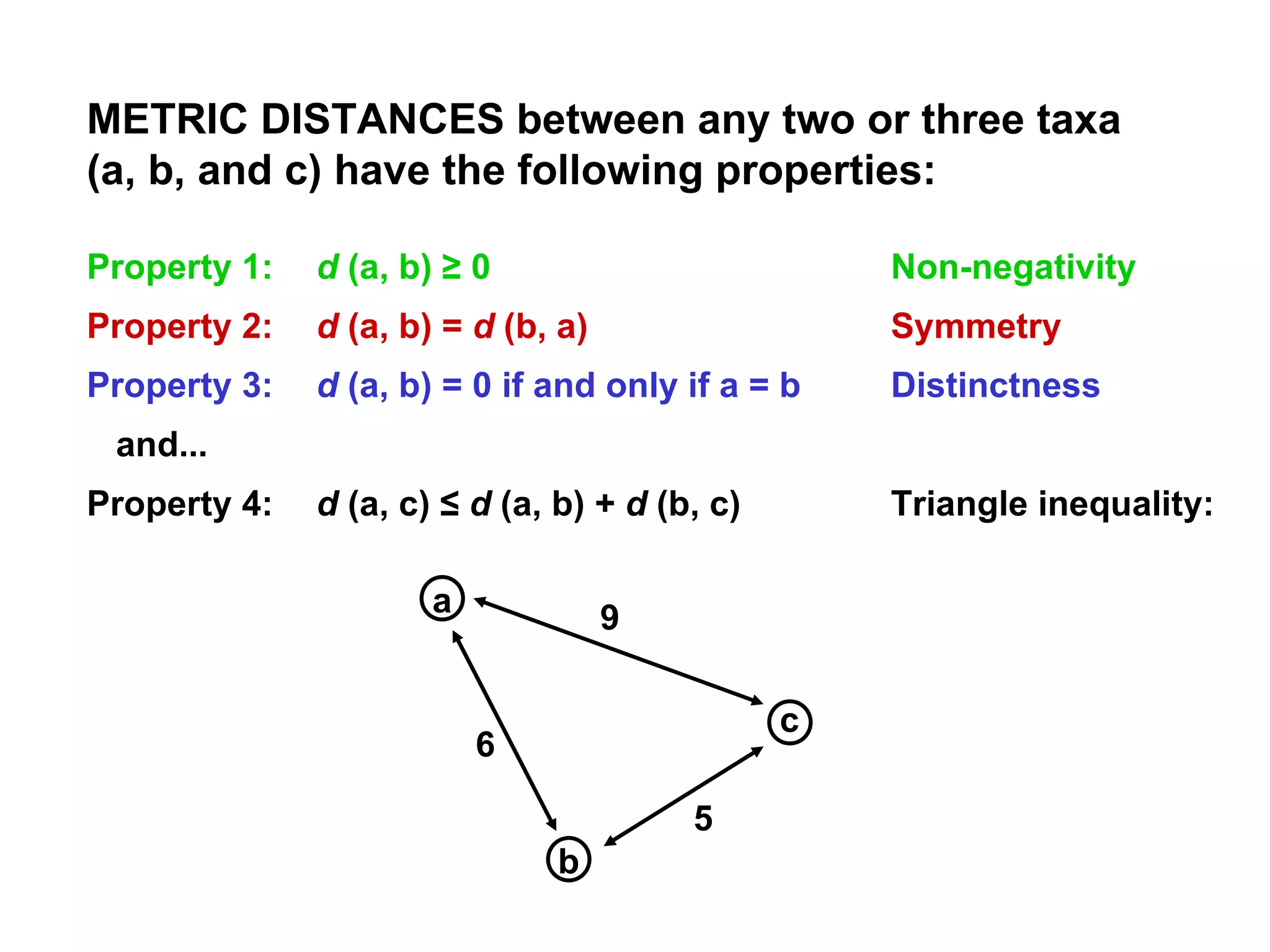
![ULTRAMETRIC DISTANCES must satisfy the previous four conditions, plus: Property 5 d (a, b) ≤ maximum [ d (a, c), d (b, c)] If distances are ultrametric, then the sequences are evolving in a perfectly clock-like manner, thus can be used in UPGMA trees and for the most precise calculations of divergence dates. Similarity = Relationship if the distances are ultrametric! This implies that the two largest distances are equal, so that they define an isosceles triangle: a b 4 6 6 c a b c 2 2 2 4](https://image.slidesharecdn.com/phylogeneticanalysis-111117220939-phpapp01/75/Phylogenetic-analysis-25-2048.jpg)
![ADDITIVE DISTANCES: Property 6: d (a, b) + d (c, d) ≤ maximum [ d (a, c) + d (b, d), d (a, d) + d (b, c)] For distances to fit into an evolutionary tree, they must be either metric or ultrametric, and they must be additive. Estimated distances often fall short of these criteria, and thus can fail to produce correct evolutionary trees.](https://image.slidesharecdn.com/phylogeneticanalysis-111117220939-phpapp01/75/Phylogenetic-analysis-26-2048.jpg)


![Parsimony methods: Optimality criterion: The ‘most-parsimonious’ tree is the one that requires the fewest number of evolutionary events ( e.g., nucleotide substitutions, amino acid replacements) to explain the sequences. Advantages: Are simple, intuitive, and logical (many possible by ‘pencil-and-paper’). Can be used on molecular and non-molecular ( e.g., morphological) data. Can tease apart types of similarity (shared-derived, shared-ancestral, homoplasy) Can be used for character (can infer the exact substitutions) and rate analysis. Can be used to infer the sequences of the extinct (hypothetical) ancestors. Disadvantages: Are simple, intuitive, and logical (derived from “Medieval logic”, not statistics!) Can be fooled by high levels of homoplasy (‘same’ events). Can become positively misleading in the “Felsenstein Zone”: [See Stewart (1993) for a simple explanation of parsimony analysis, and Swofford et al. (1996) for a detailed explanation of various parsimony methods.]](https://image.slidesharecdn.com/phylogeneticanalysis-111117220939-phpapp01/75/Phylogenetic-analysis-32-2048.jpg)






![Highly Recommended Programs for Phylogenetic Inference and Evolutionary Analysis Swofford, D.M. (1998) PAUP* 4: Phylogenetic Analysis Using Parsimony (*and Other Methods) . Sinauer Associates, Sunderland, MA. This is the most versatile and user-friendly phylogenetic analysis package currently available. PAUP* has parsimony, likelihood, and distance methods. It is sold for a nominal cost. Available for several platforms; the PowerMac version is fast and menu-driven. Maddison, D.R & Maddison, W.P. (2000) MacClade 4: Analysis of Phylogeny and Character Evolution. Sinauer Associates, Sunderland, MA. This is a versatile and user-friendly program that aids greatly in character analysis of molecular (and other) data. One can readily ‘build’ trees by click-and-drop methods, and save them for further analyses. Available for Macintosh and MacOS emulators. Fun! Yang, Z. (1998) PAML: Phylogenetic Analysis using Maximum Likelihood. [Available from the author or online.] This is the scientifically best program available for testing alternative models of molecular evolution in a phylogenetic ML framework. Is user-hostile, but worth the effort. Available for several platforms.](https://image.slidesharecdn.com/phylogeneticanalysis-111117220939-phpapp01/75/Phylogenetic-analysis-39-2048.jpg)


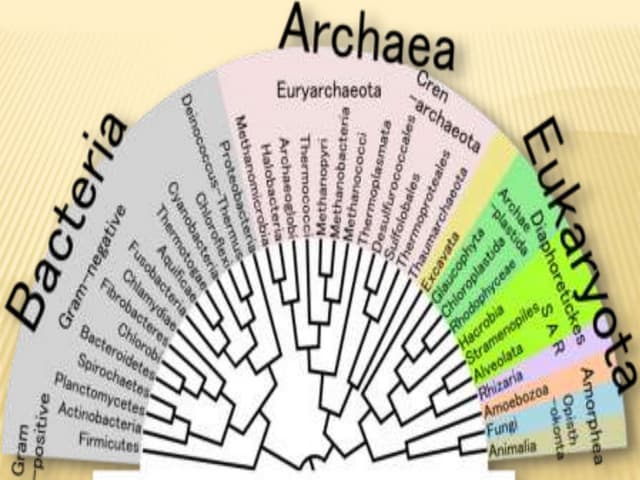
This document provides an overview of phylogenetic analysis, including: 1) Phylogenetic analysis involves inferring evolutionary relationships between taxa by building phylogenetic trees and analyzing character evolution. 2) Phylogenetic trees show the branching patterns and relationships between taxa, with internal nodes representing hypothetical ancestors. 3) Phylogenetic analysis can provide insights into questions like human evolution, disease transmission, and the origins of genetic elements.
Overview of the lecture by Caro-Beth Stewart introducing phylogenetic analysis.
Phylogenetic analysis involves phylogeny inference (tree building) and character/rate analysis for understanding evolutionary relationships.
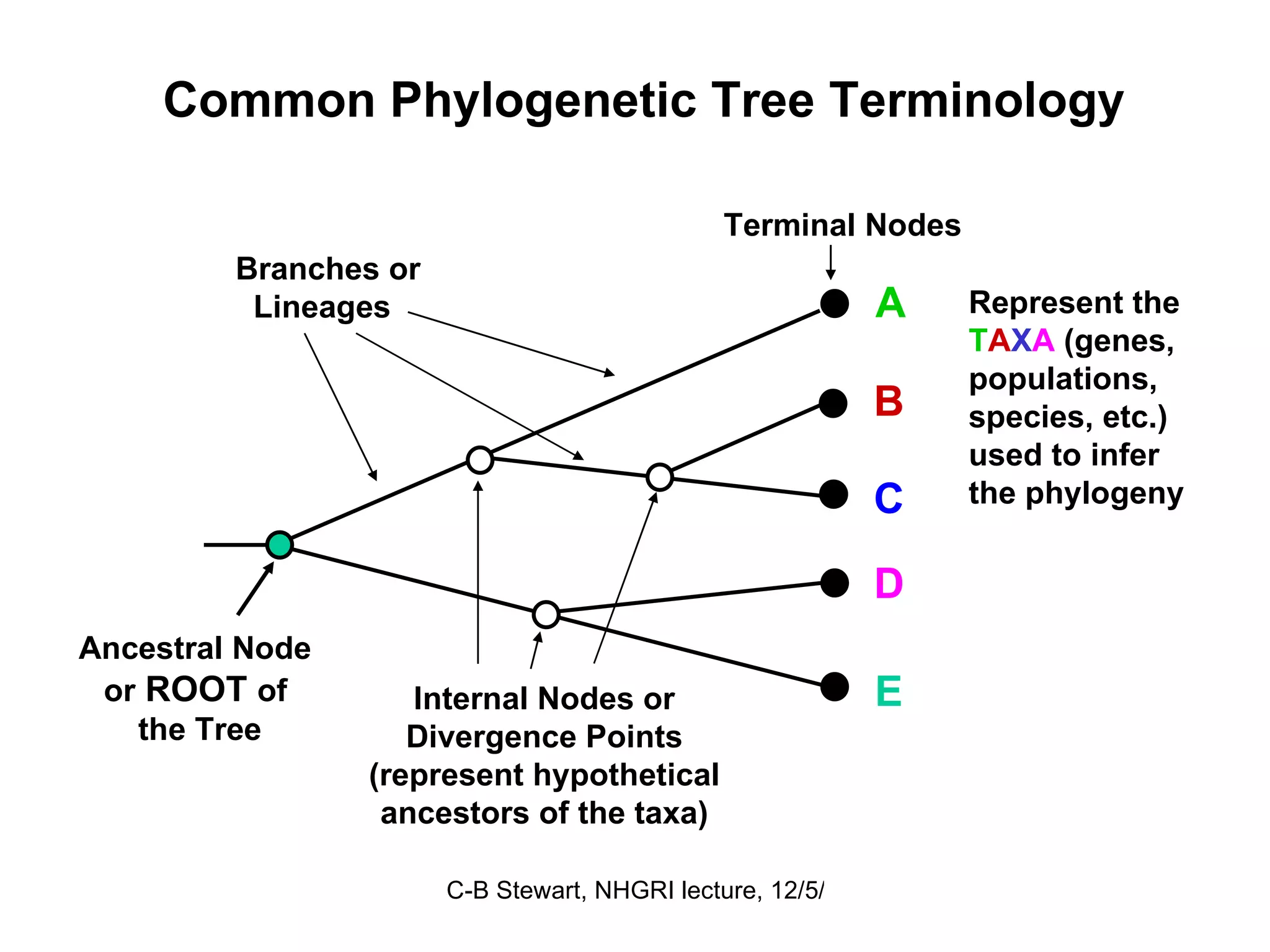
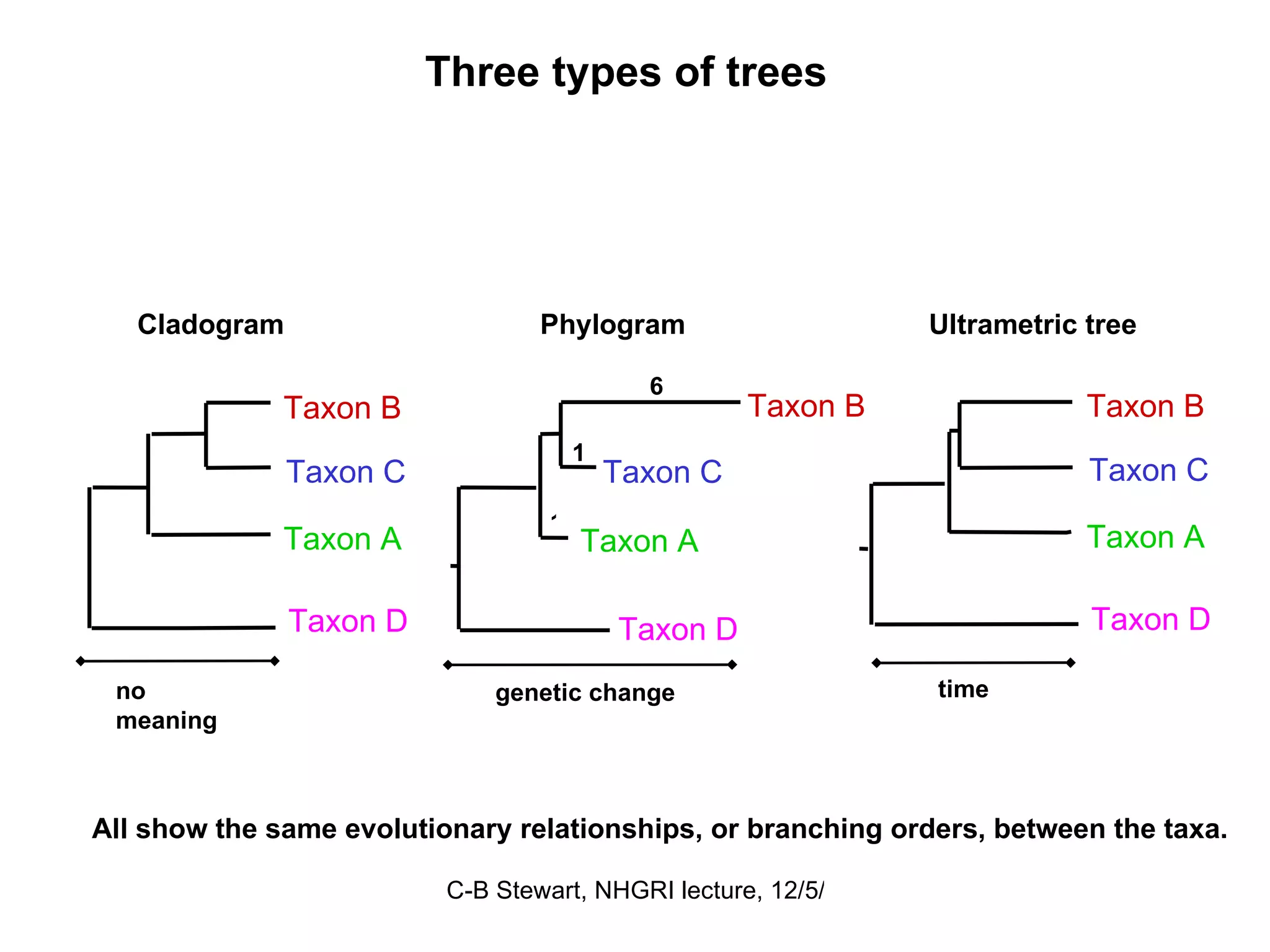
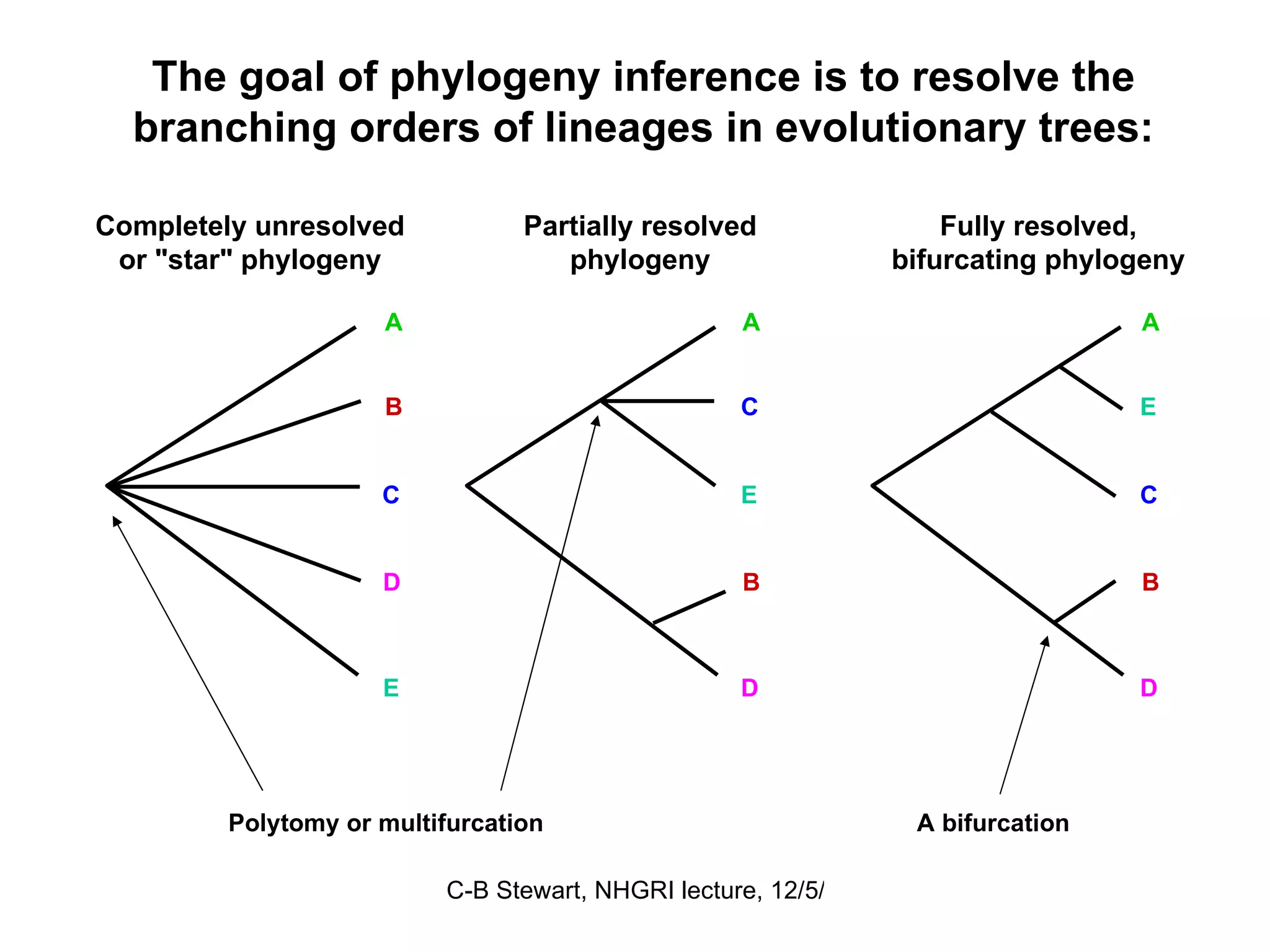
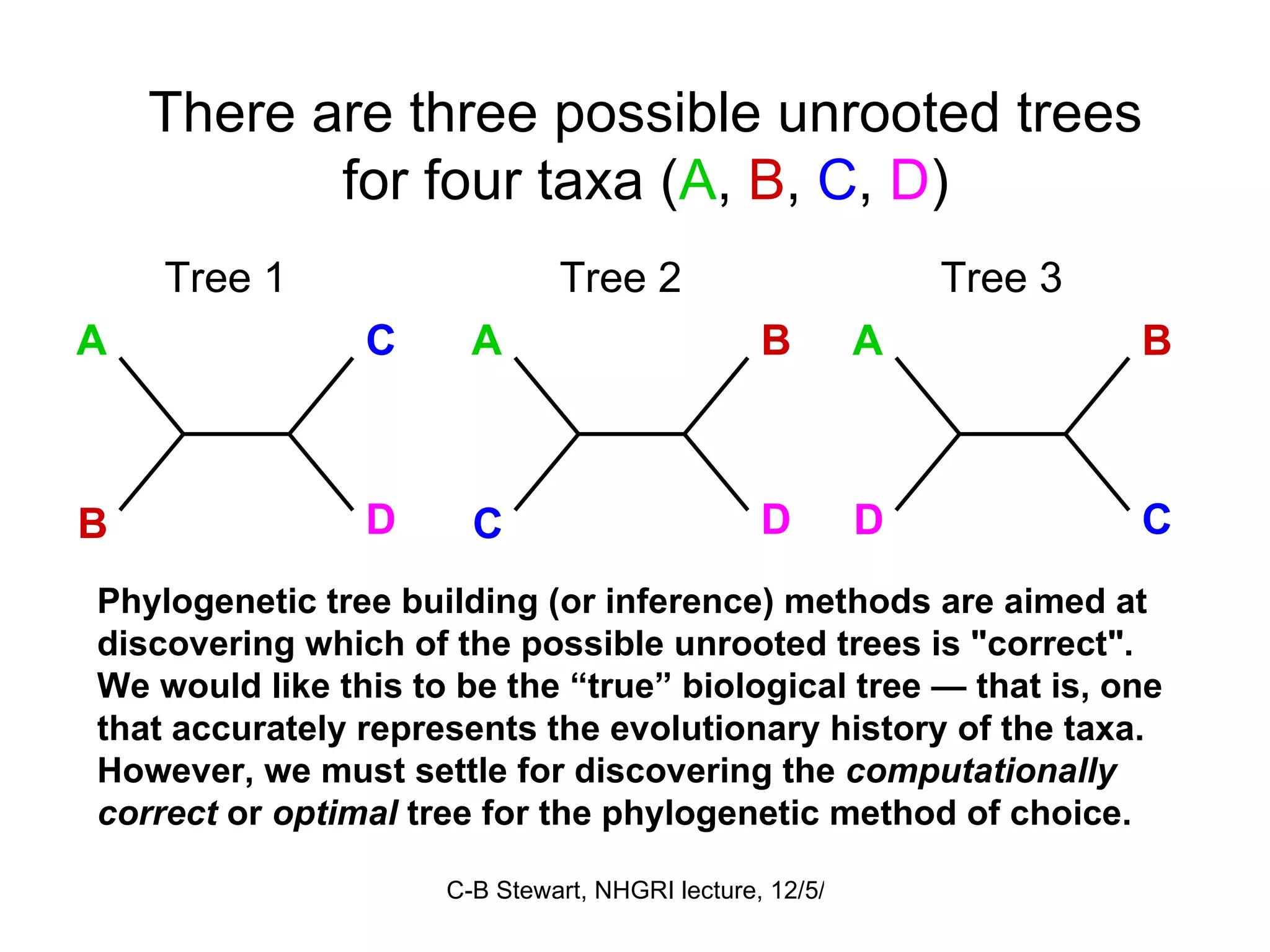
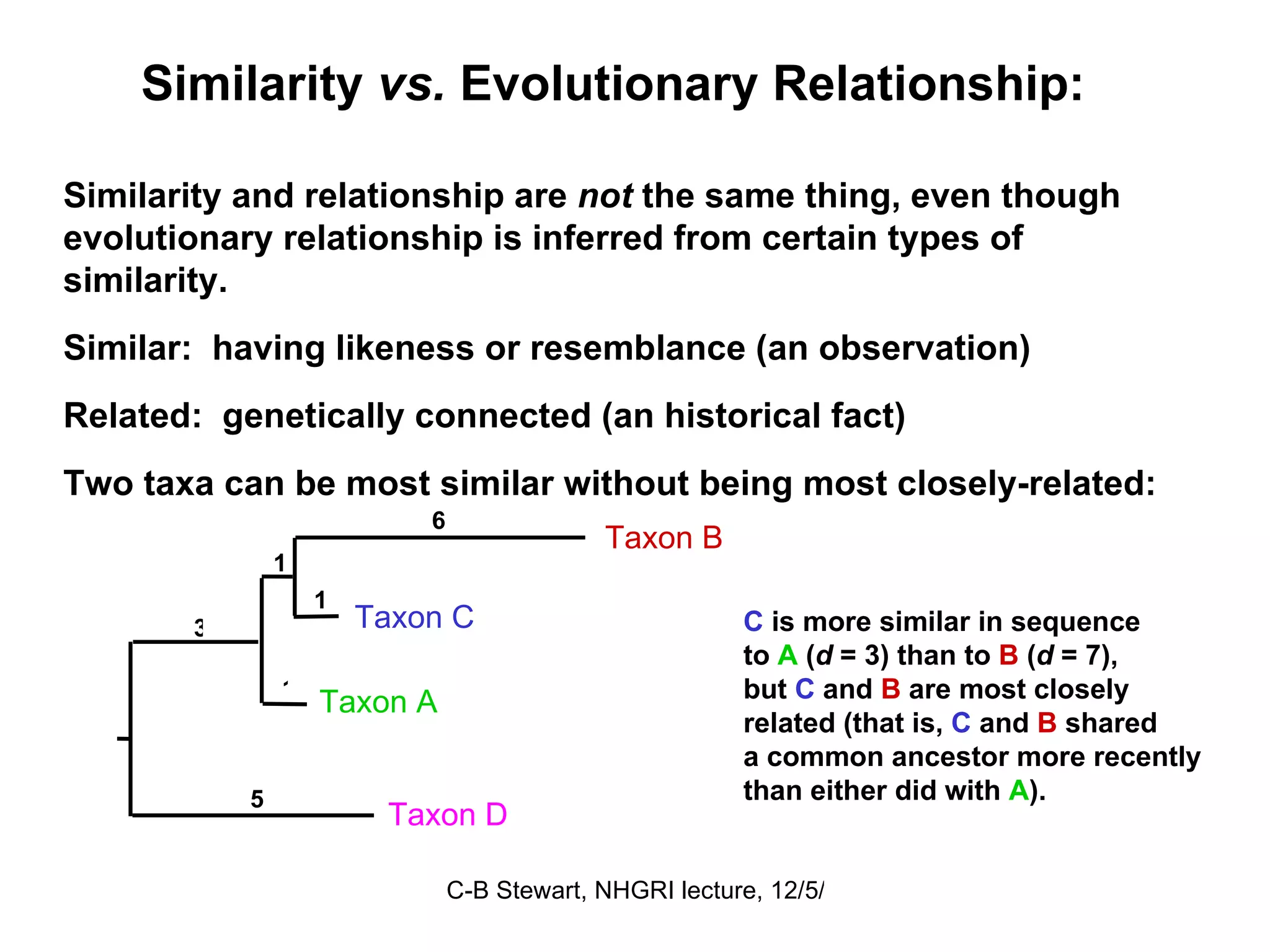
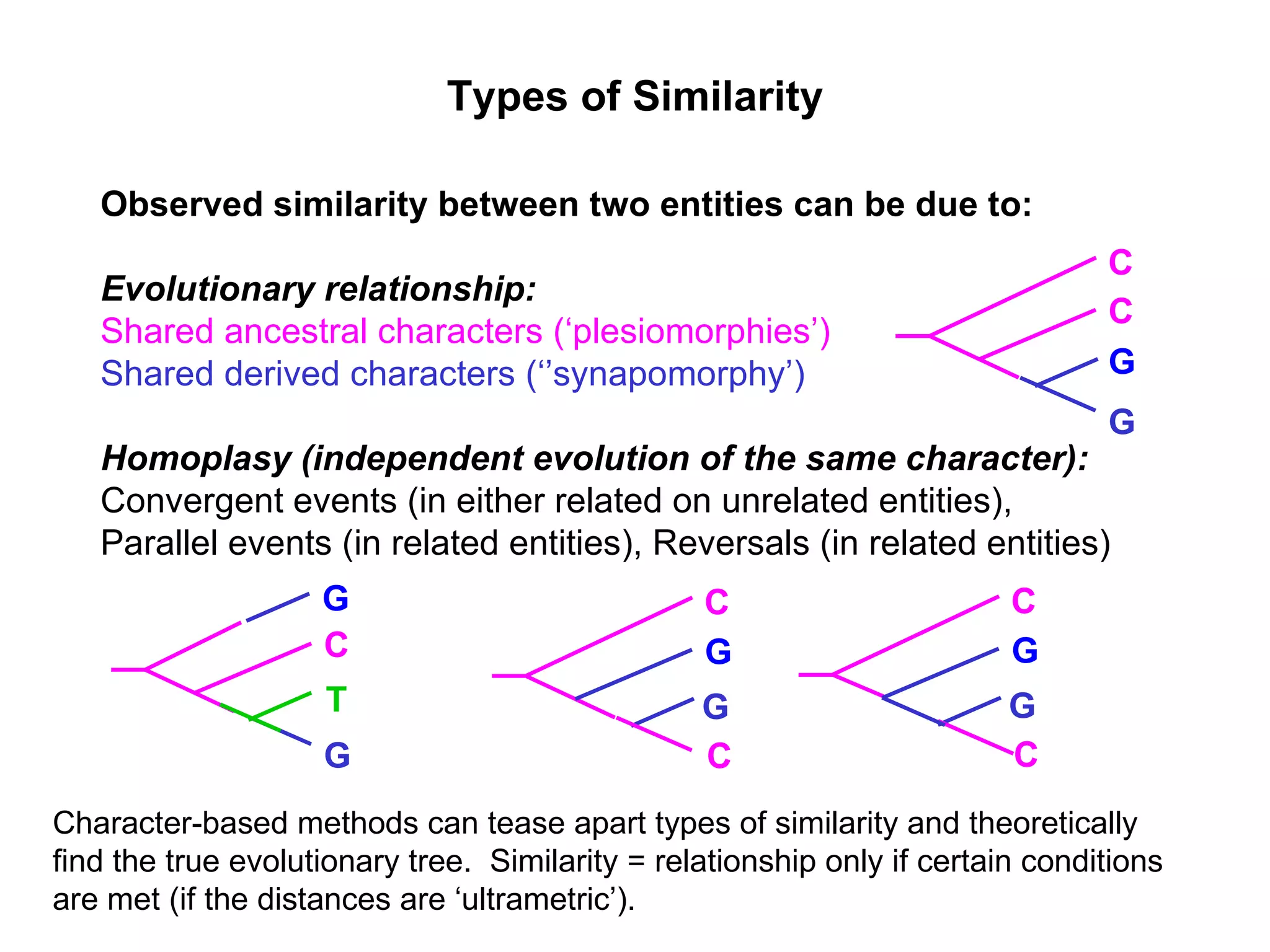
Explanation of common terms in phylogenetics including ancestral nodes, internal nodes, and different types of trees (cladogram, phylogram, ultrametric).
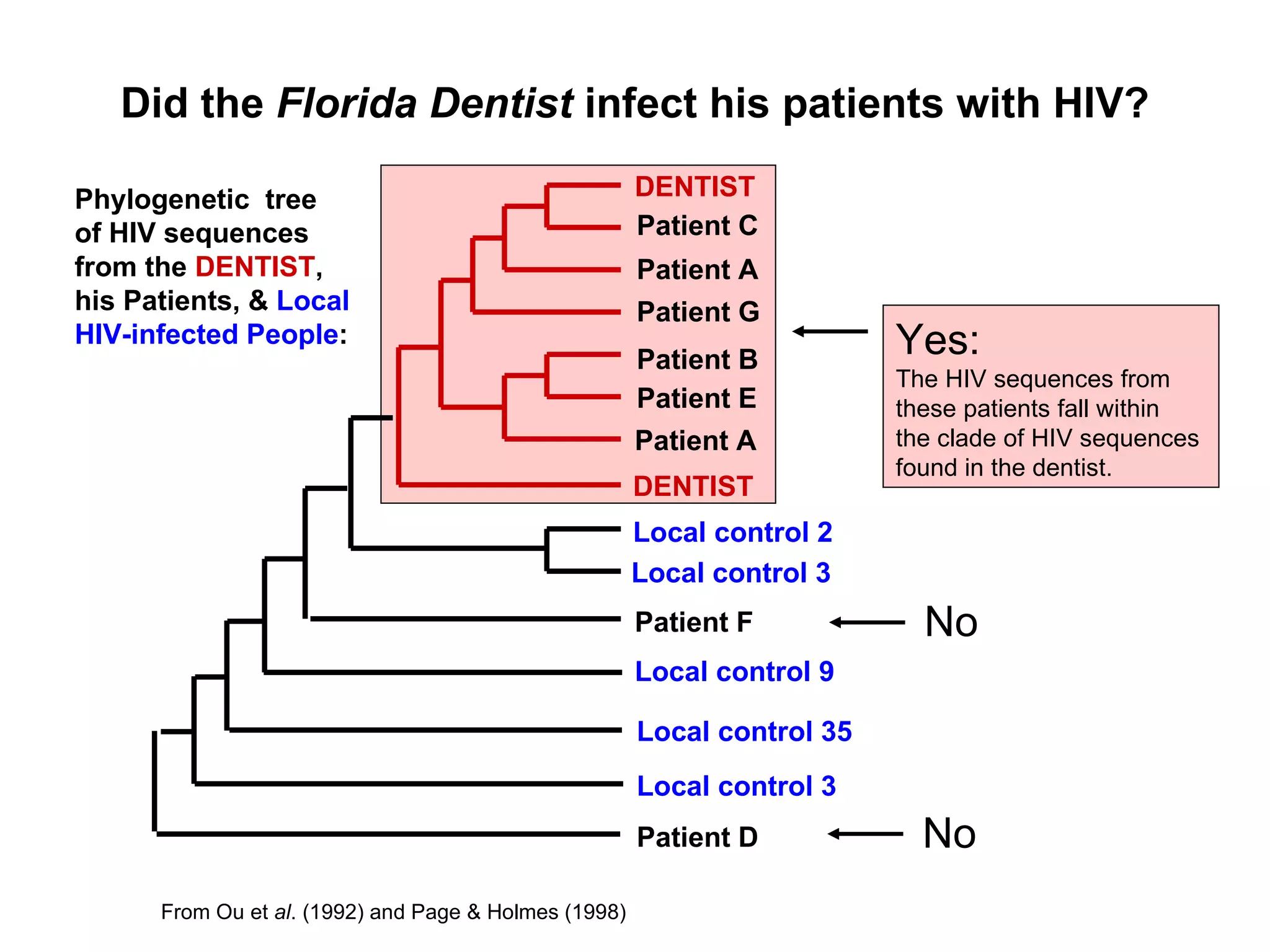
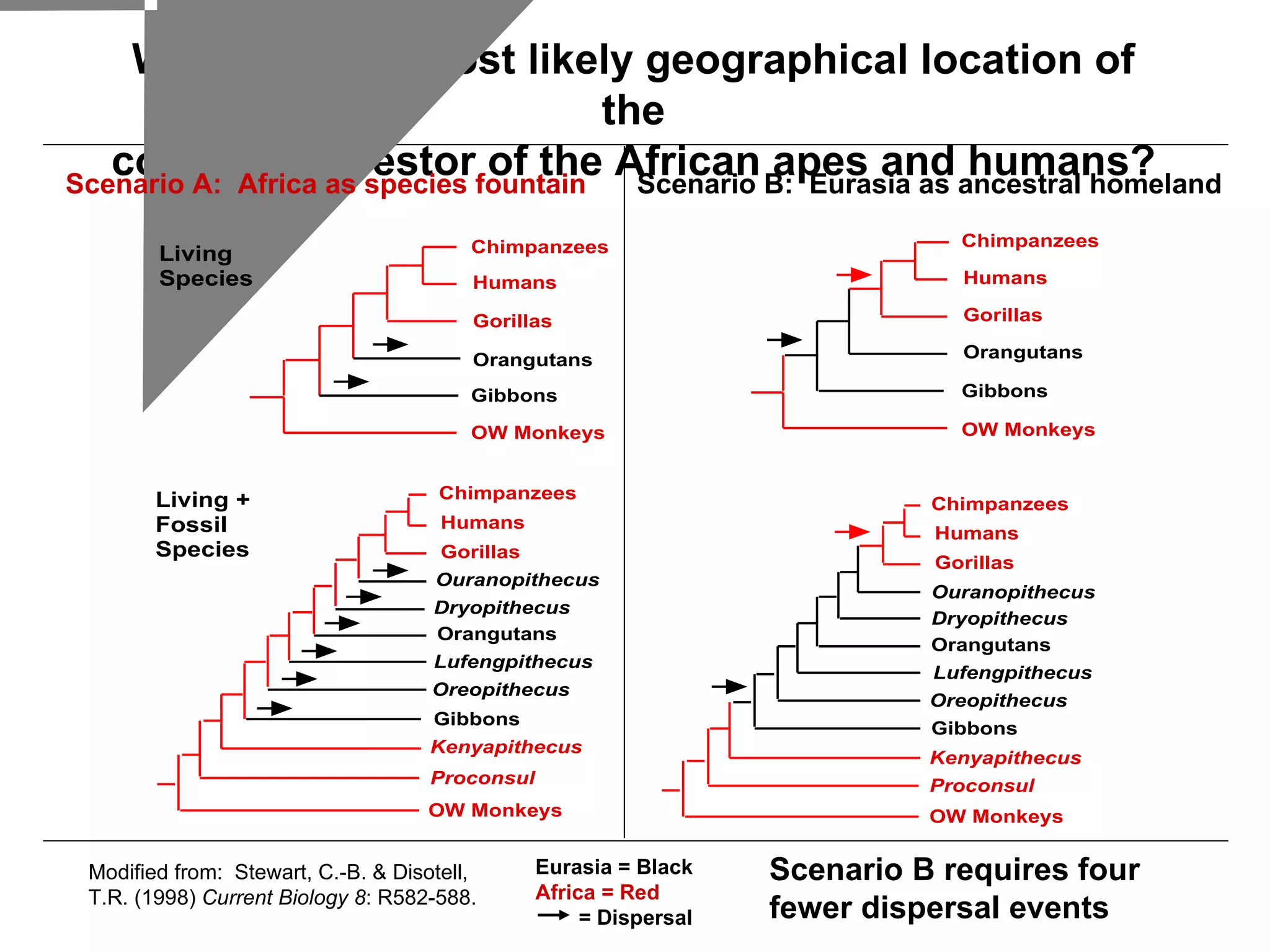
Examples of phylogenetic trees in determining species relationships, infections (HIV), and understanding human evolution with a focus on character analysis.
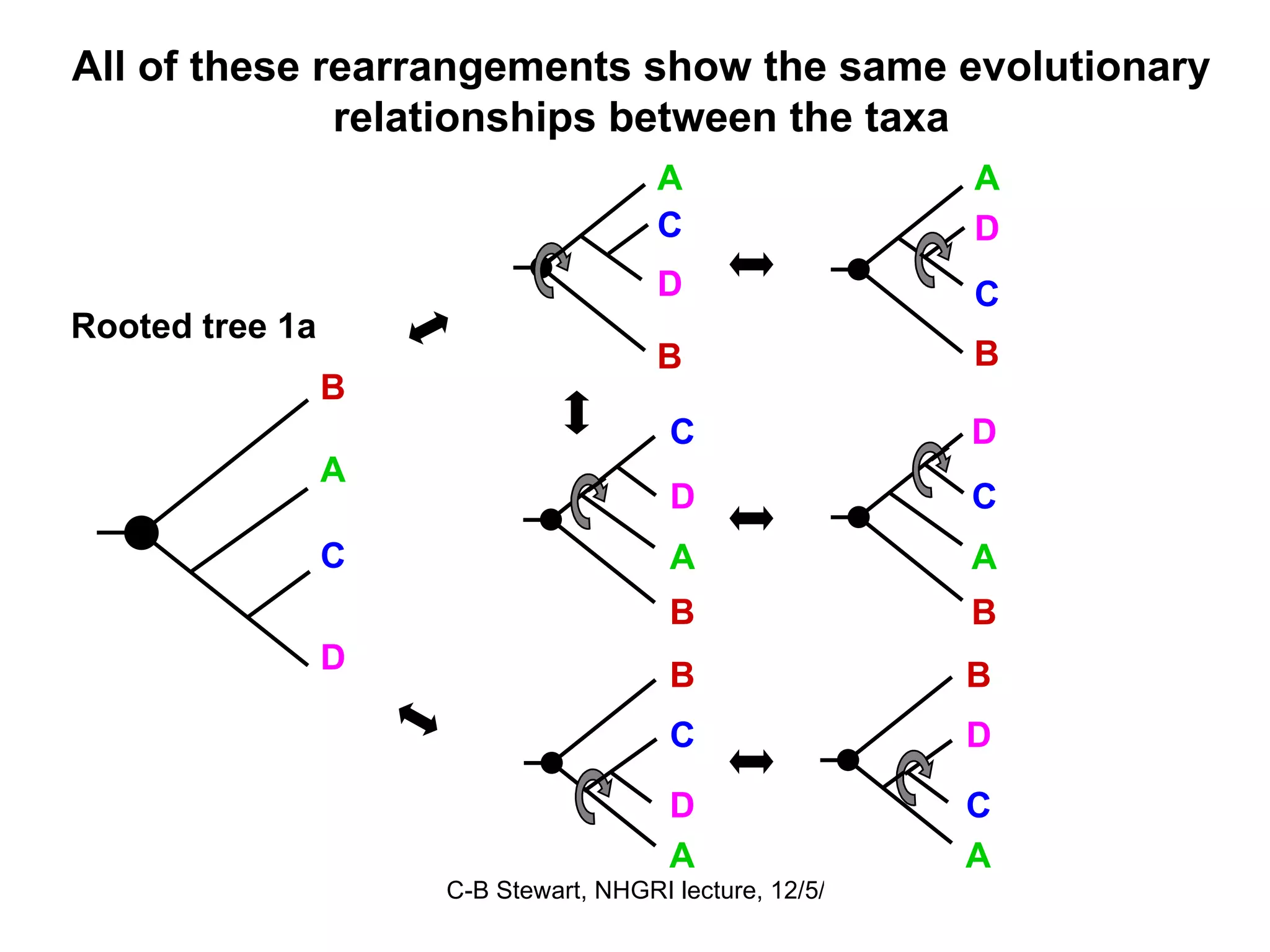
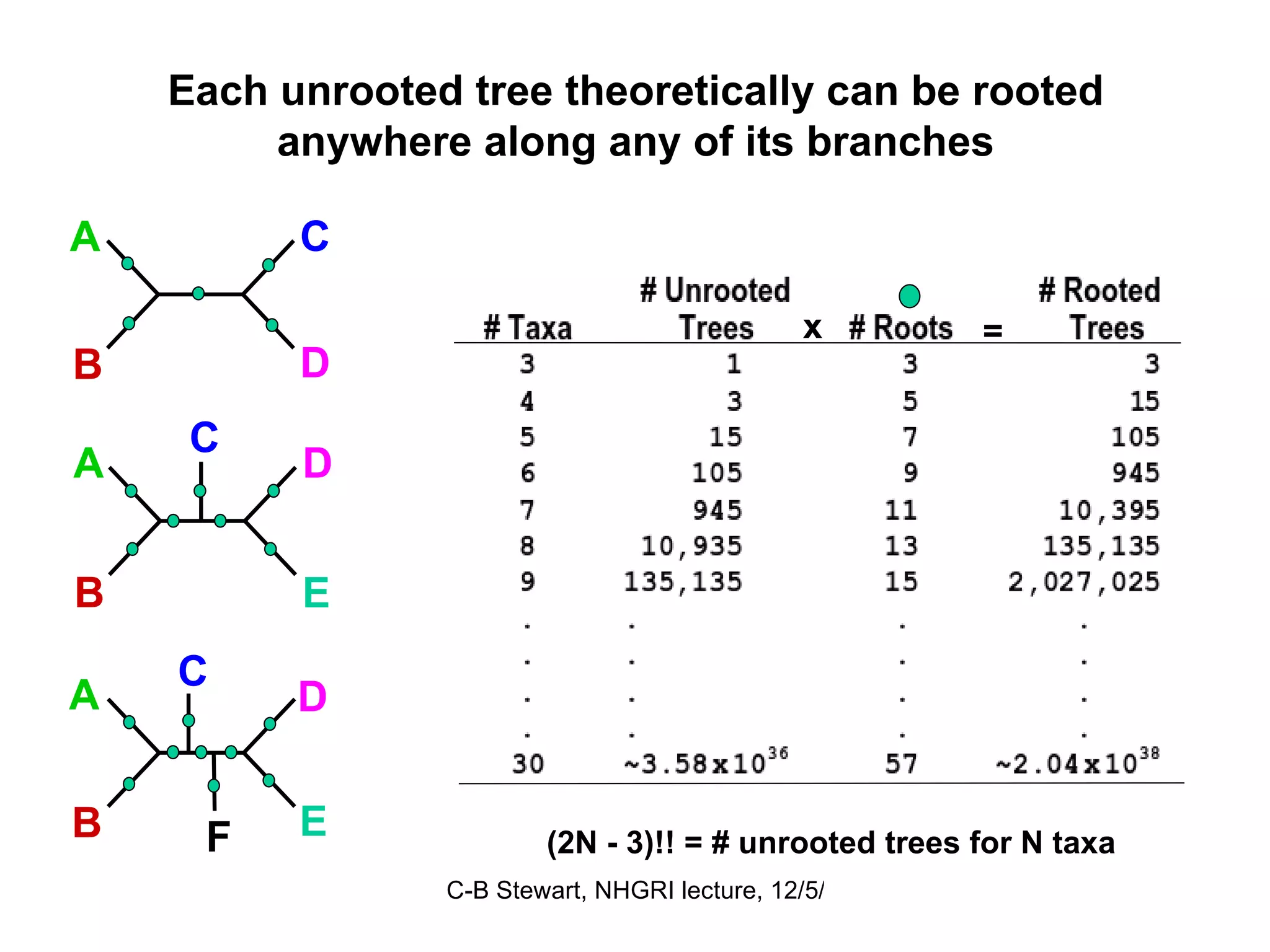
Inferred dispersal patterns and challenges in tree resolutions, including various types of phylogenetic trees and rooting methodologies.
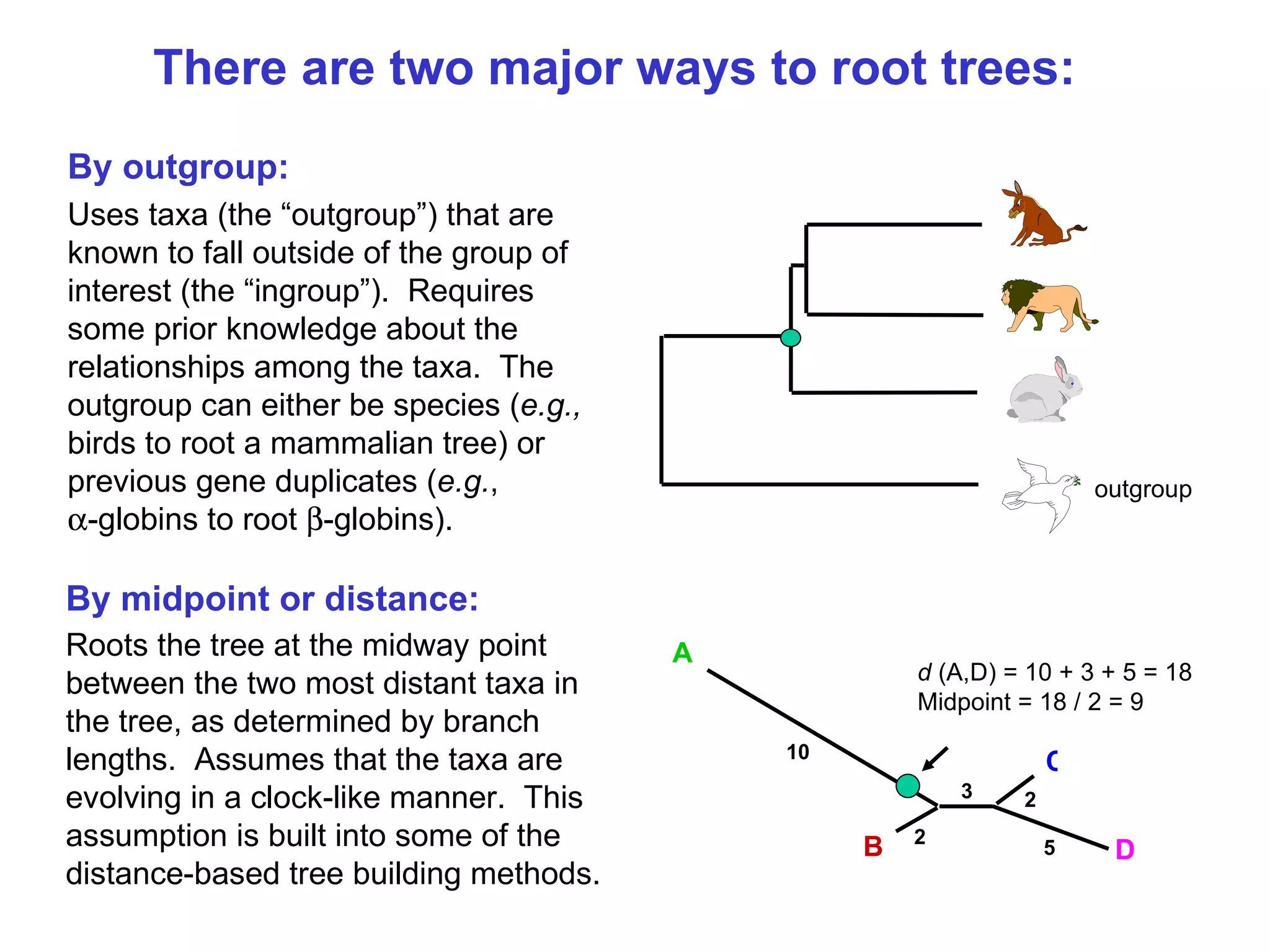
Different rooting techniques for trees, including outgroup and midpoint rooting, and their implications for evolutionary inference.
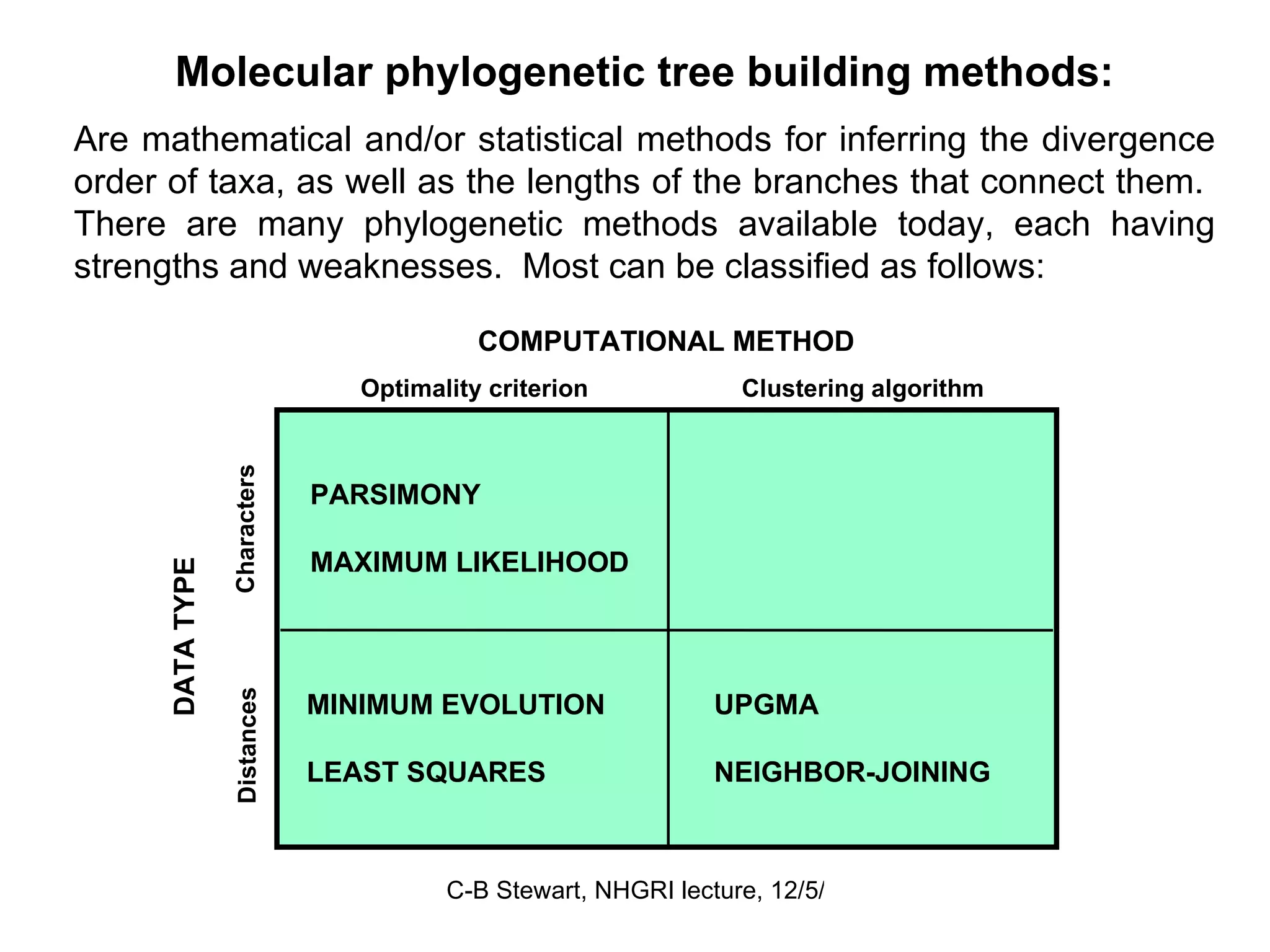
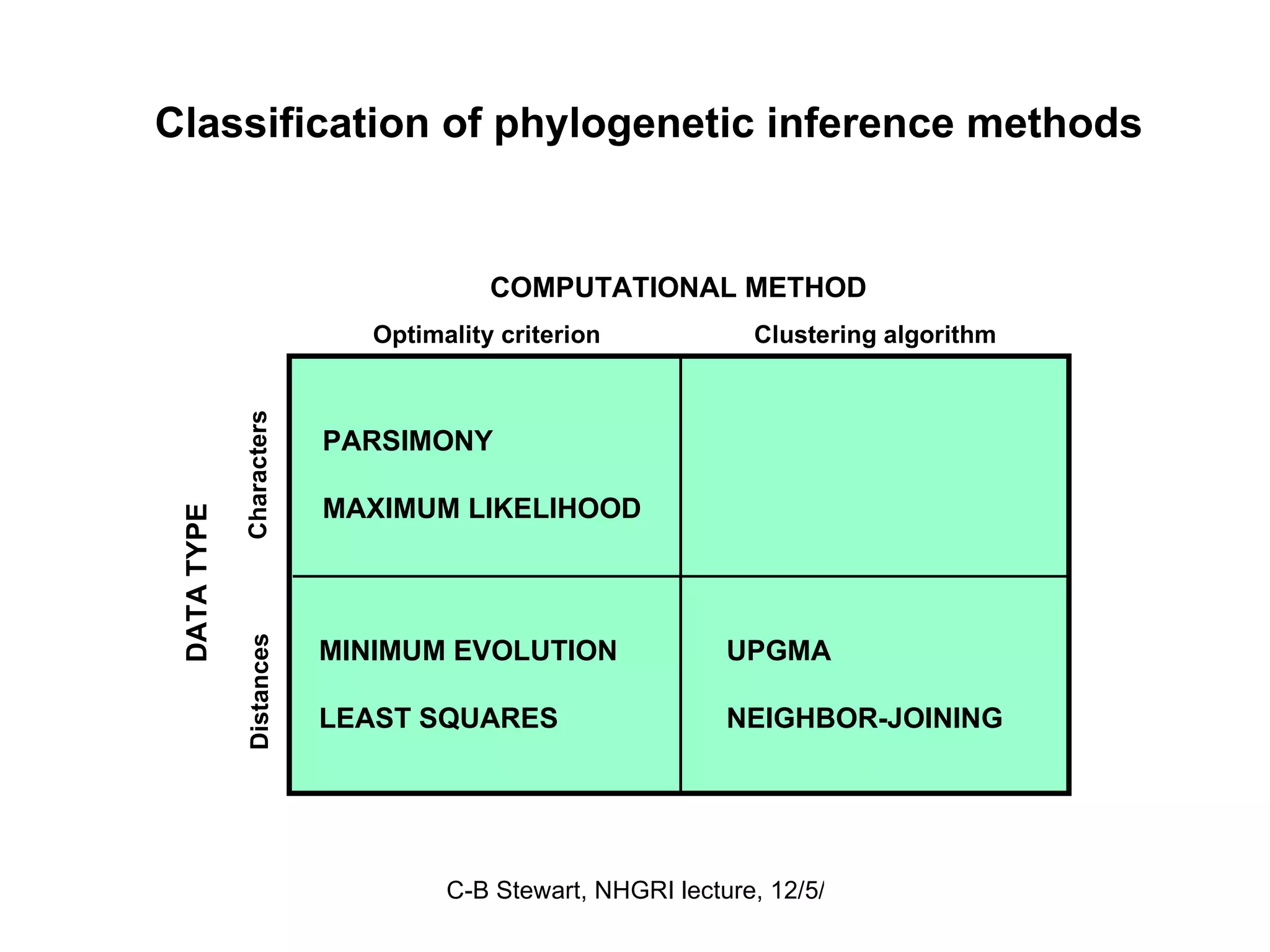
Overview of computational methods in phylogenetic analysis such as parsimony and maximum likelihood, discussing their advantages and disadvantages.
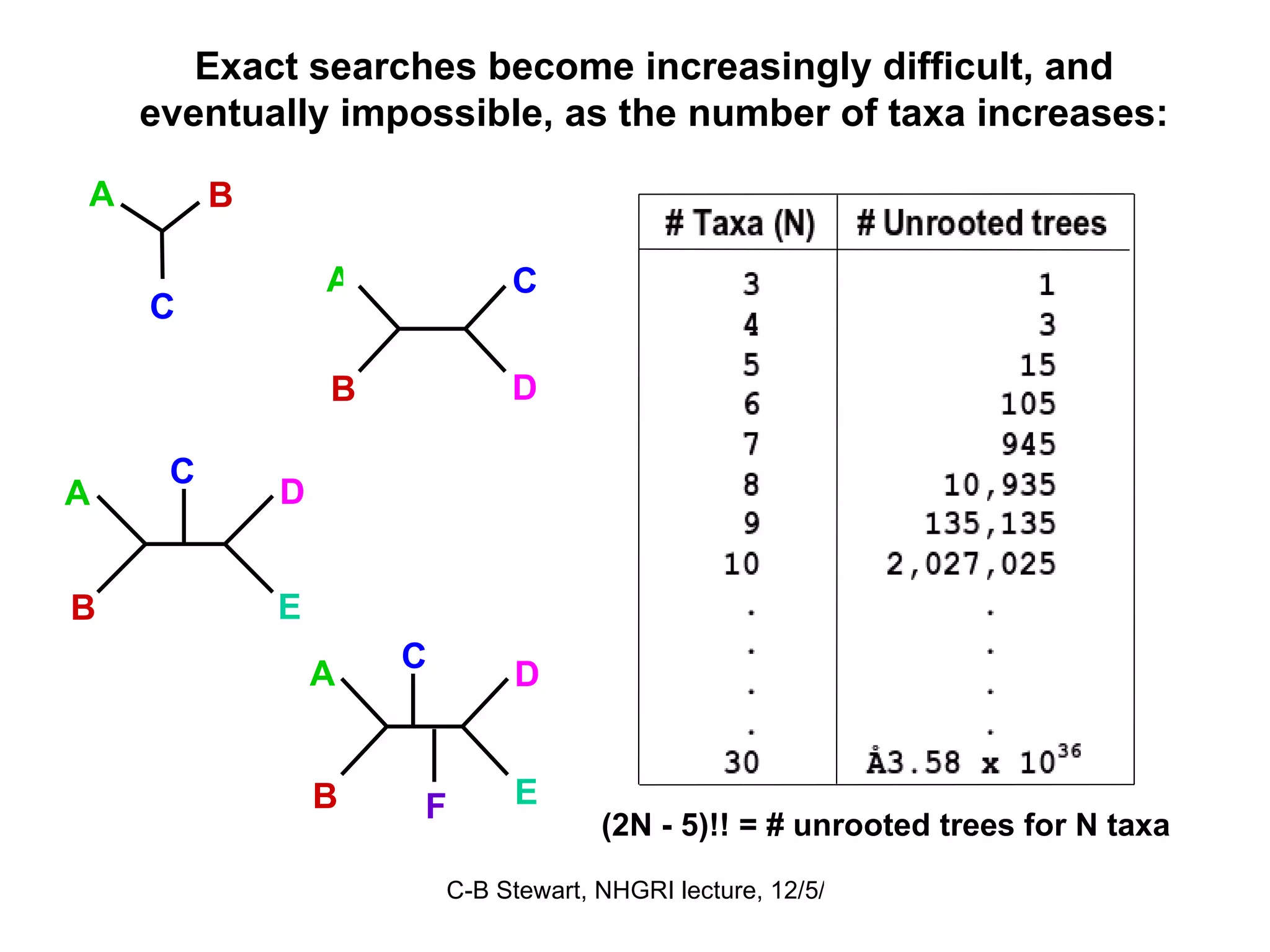
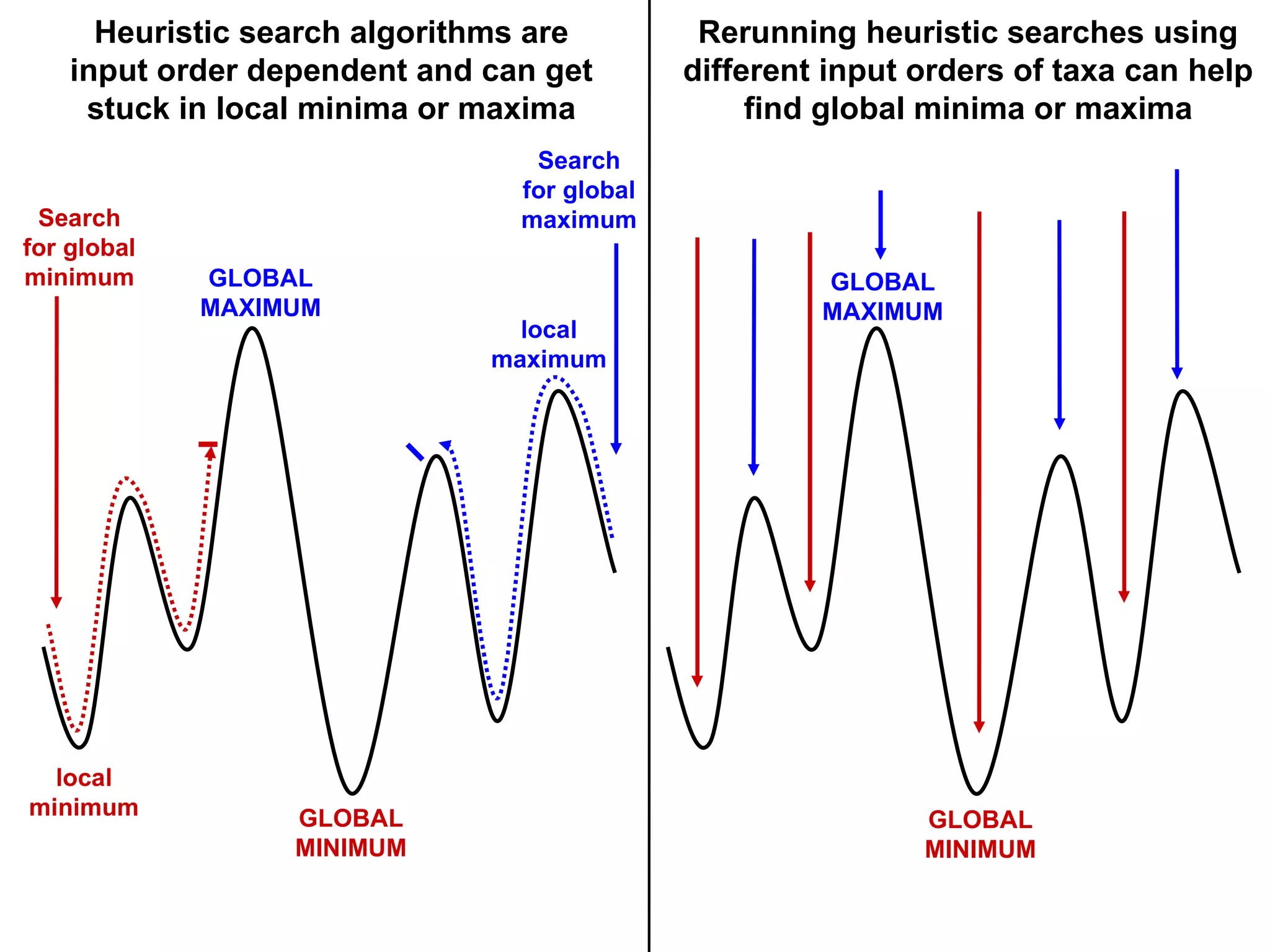
Challenges in finding optimal trees, types of search algorithms, and performance comparisons of phylogenetic methods including clustering and optimality.
Suggested literature and software tools for phylogenetic analysis, character analysis, and statistical tests comparing trees.
End of the presentation or potential for demonstrations.