Download as PDF, PPTX








































![Using BWA
bwa index [-a bwtsw|div|is] [-c] <in.fasta>
-a STR BWT construction algorithm: bwtsw or is
bwtsw for human size genome, is for smaller genomes
bwa aln [options] <prefix> <in.fq>
Align each single end fastq file individually
Various options to change the alignment parameters/scoring matrix/seed
length
bwa sampe [options] <prefix> <in1.sai> <in2.sai>
<in1.fq> <in2.fq>
sai files produced by aln step
Produces SAM output
bwa samse [-n max_occ] <prefix> <in.sai> <in.fq>
Unpaired reads – produces SAM output
Use samtools to manipulate SAM file or convert to BAM
samtools view –bS in.sam > out.bam
Thomas Keane 9th European Conference on Computational Biology 26th September, 2010](https://image.slidesharecdn.com/eccb-tutorial-thomas1-101009130915-phpapp01/75/ECCB-2010-Next-gen-sequencing-Tutorial-69-2048.jpg)
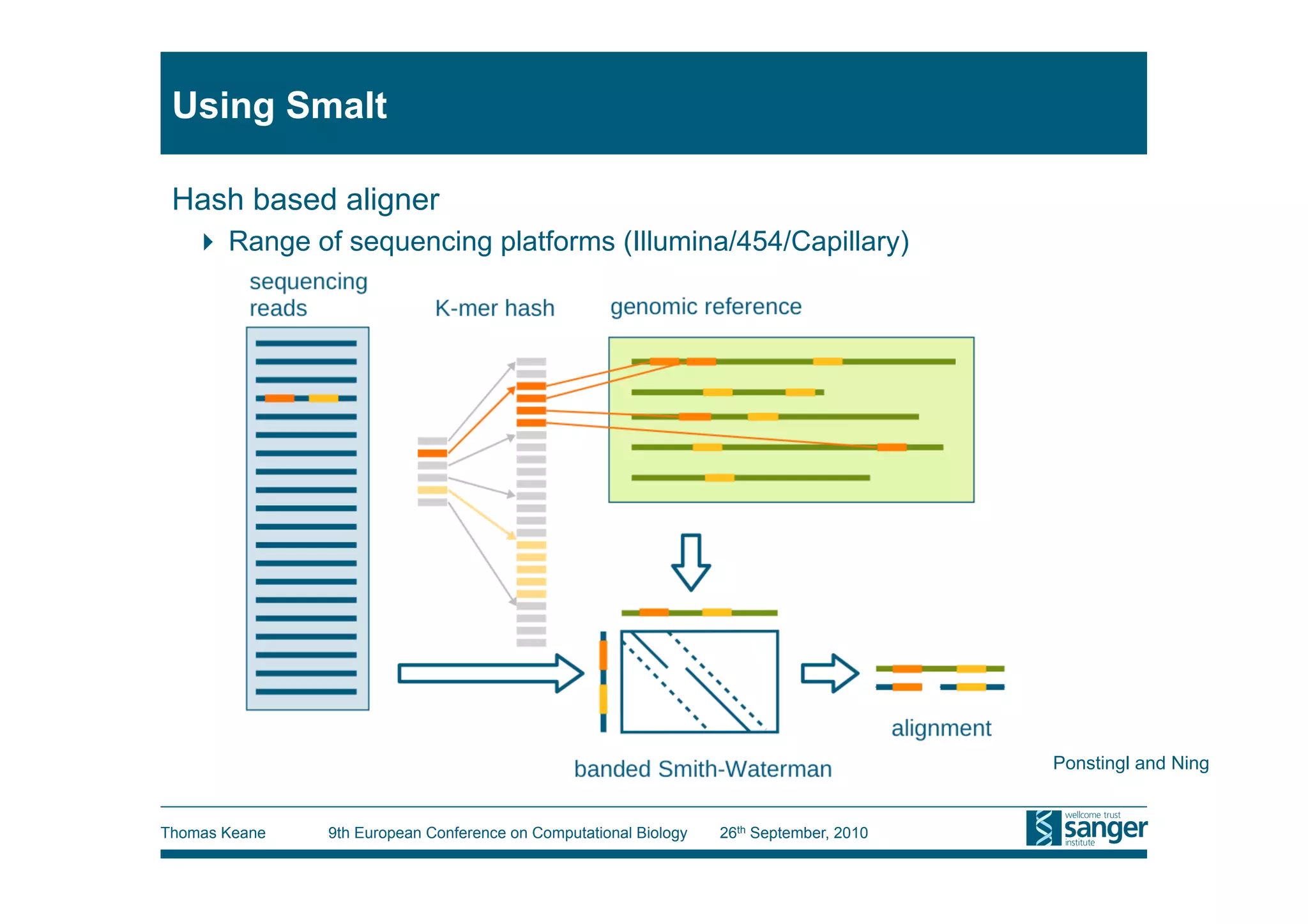
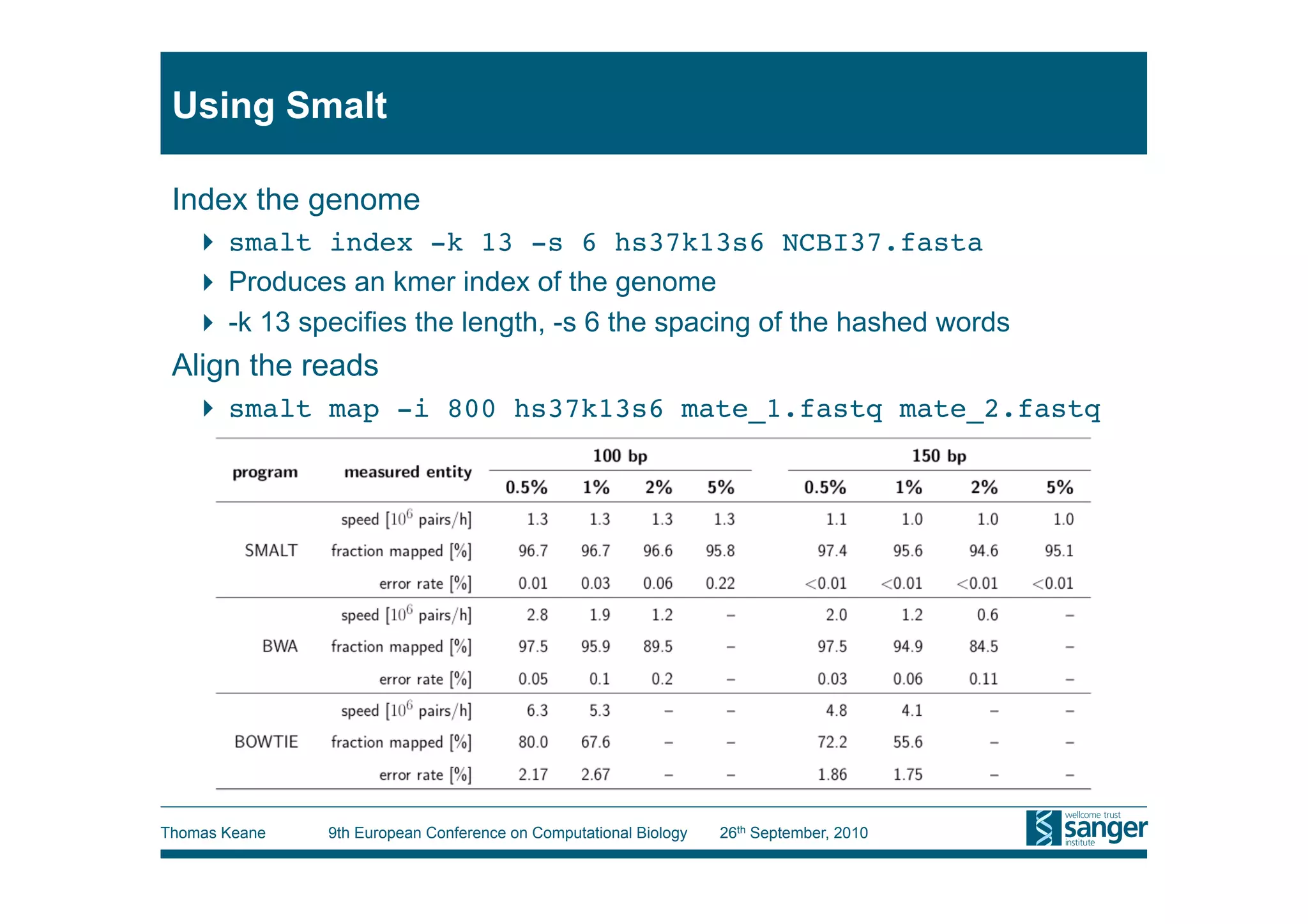




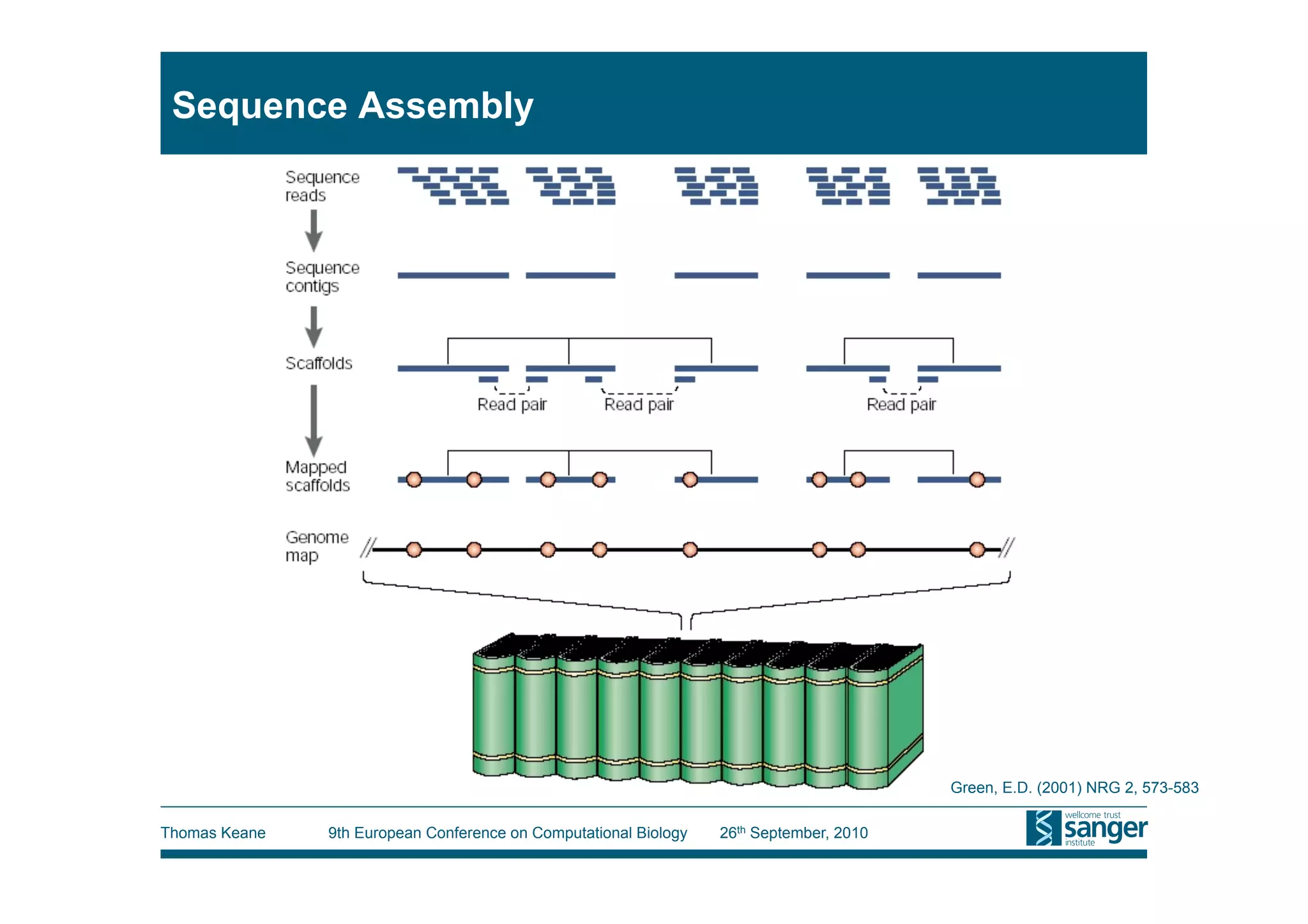

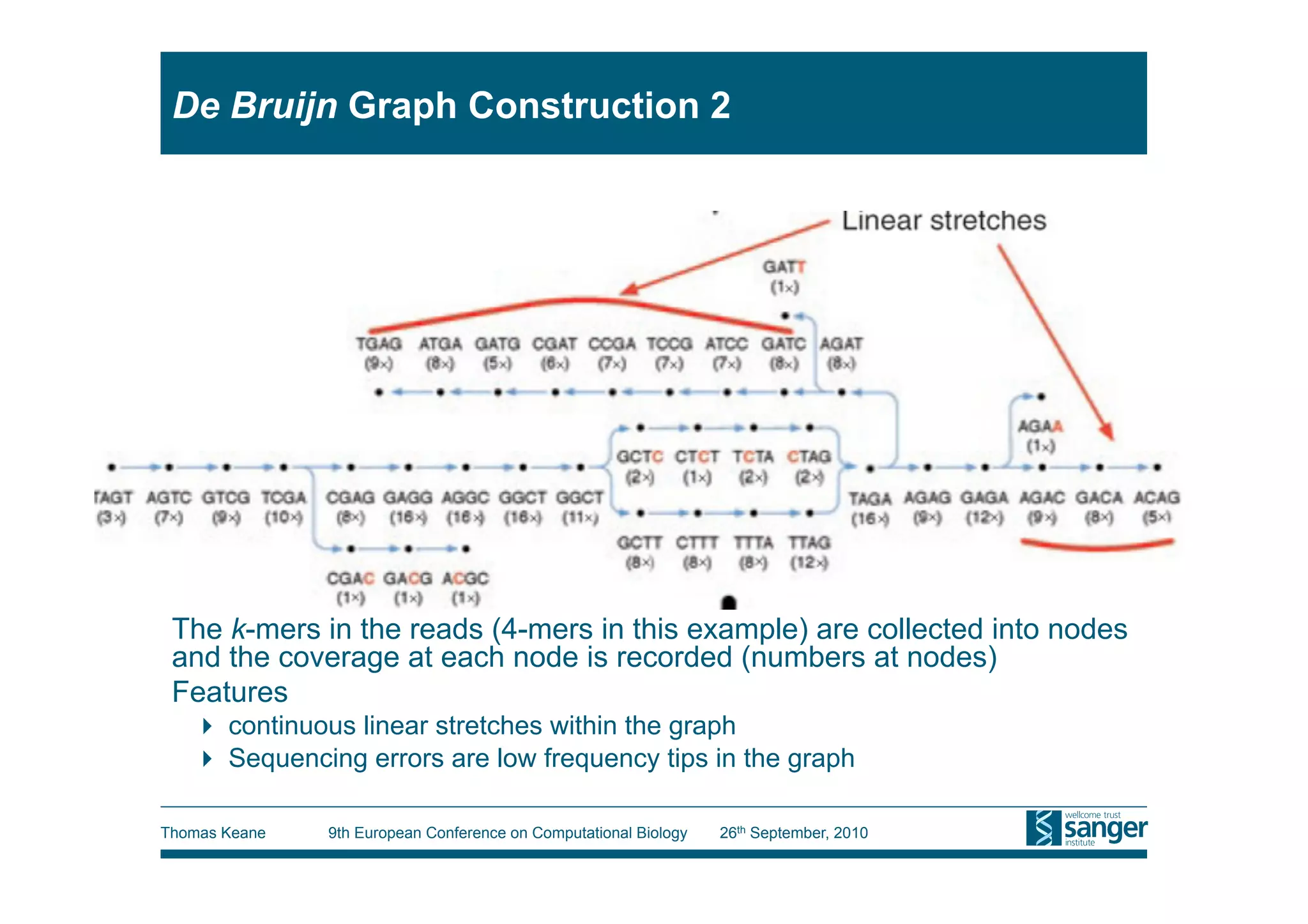
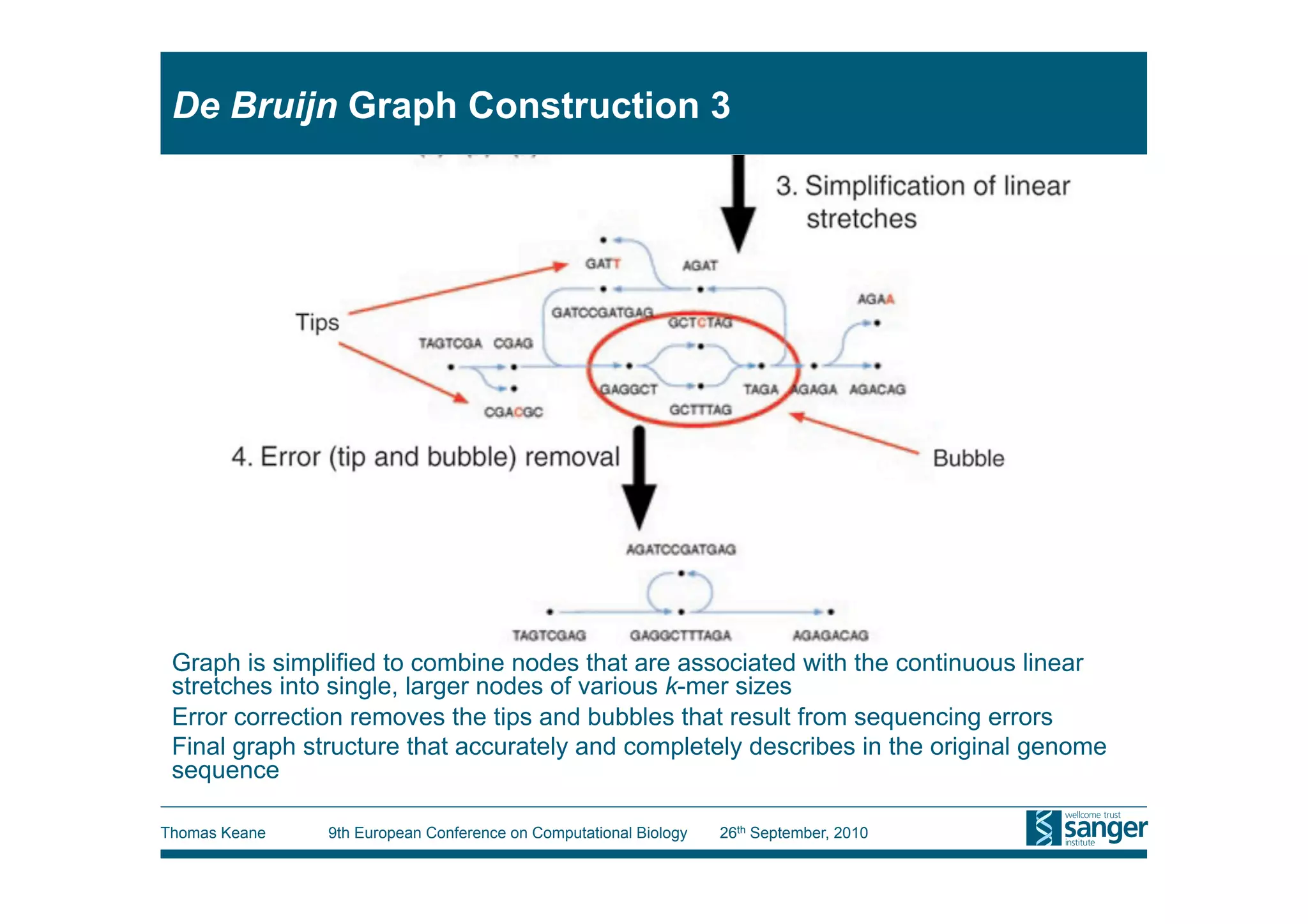


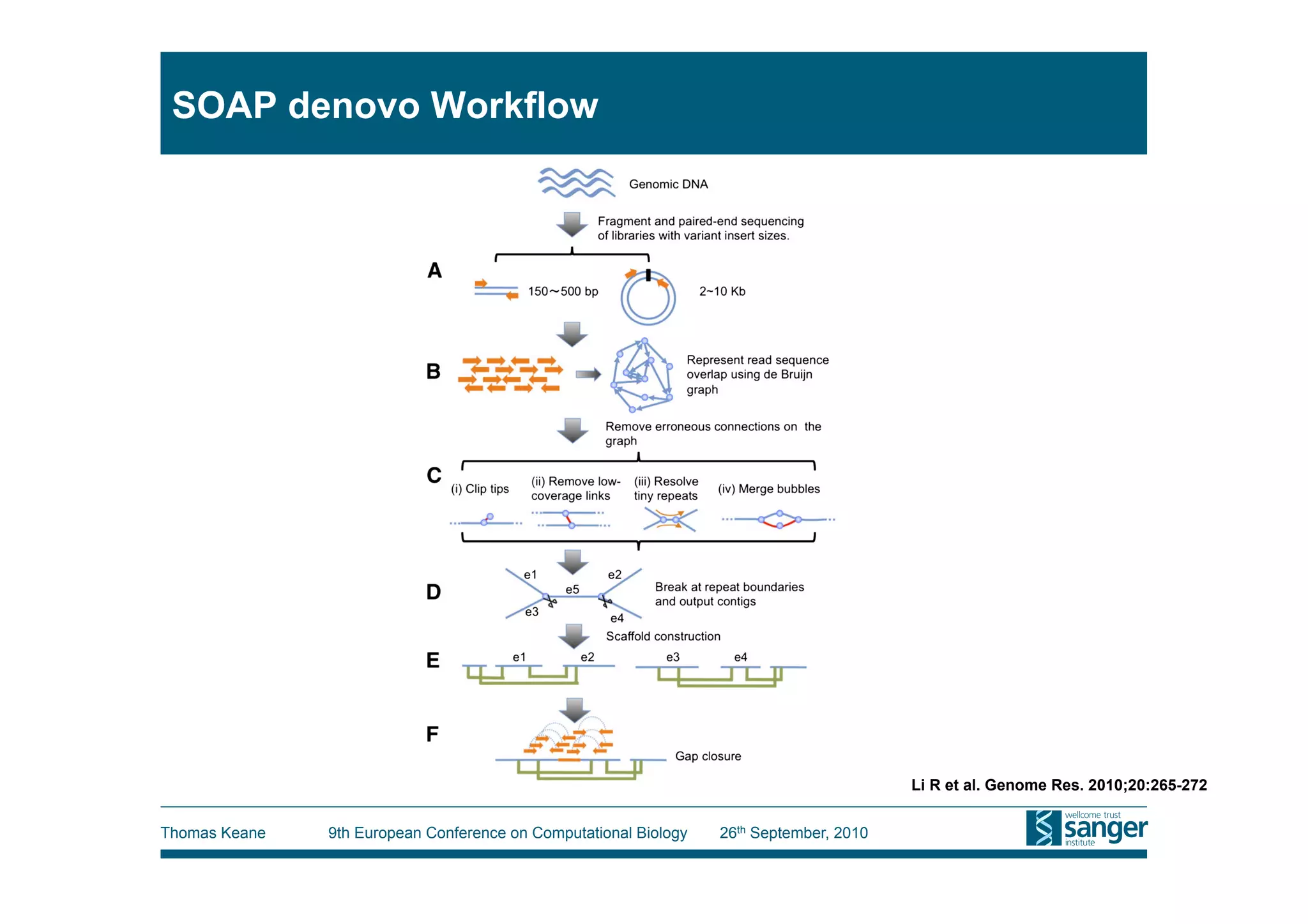
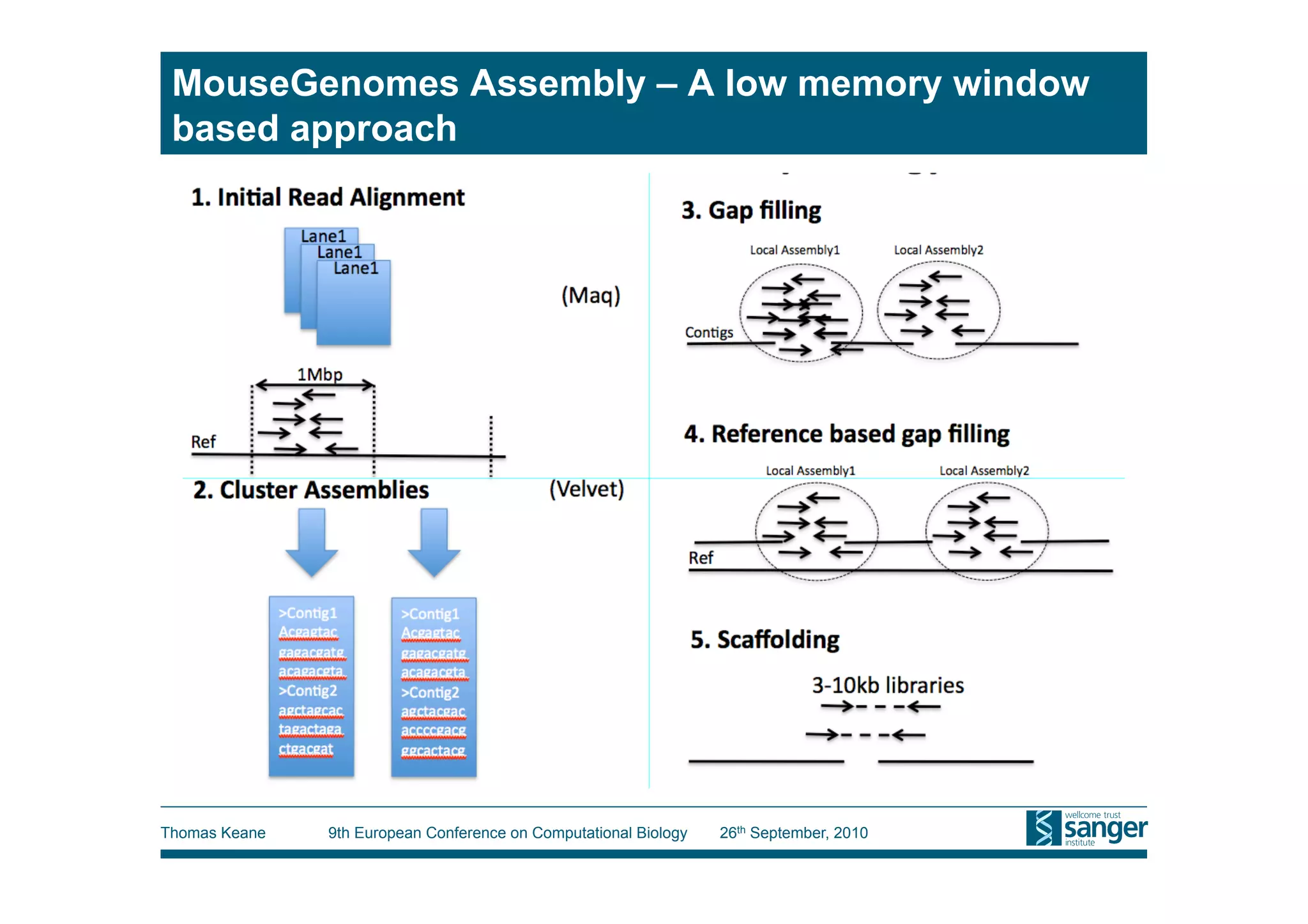

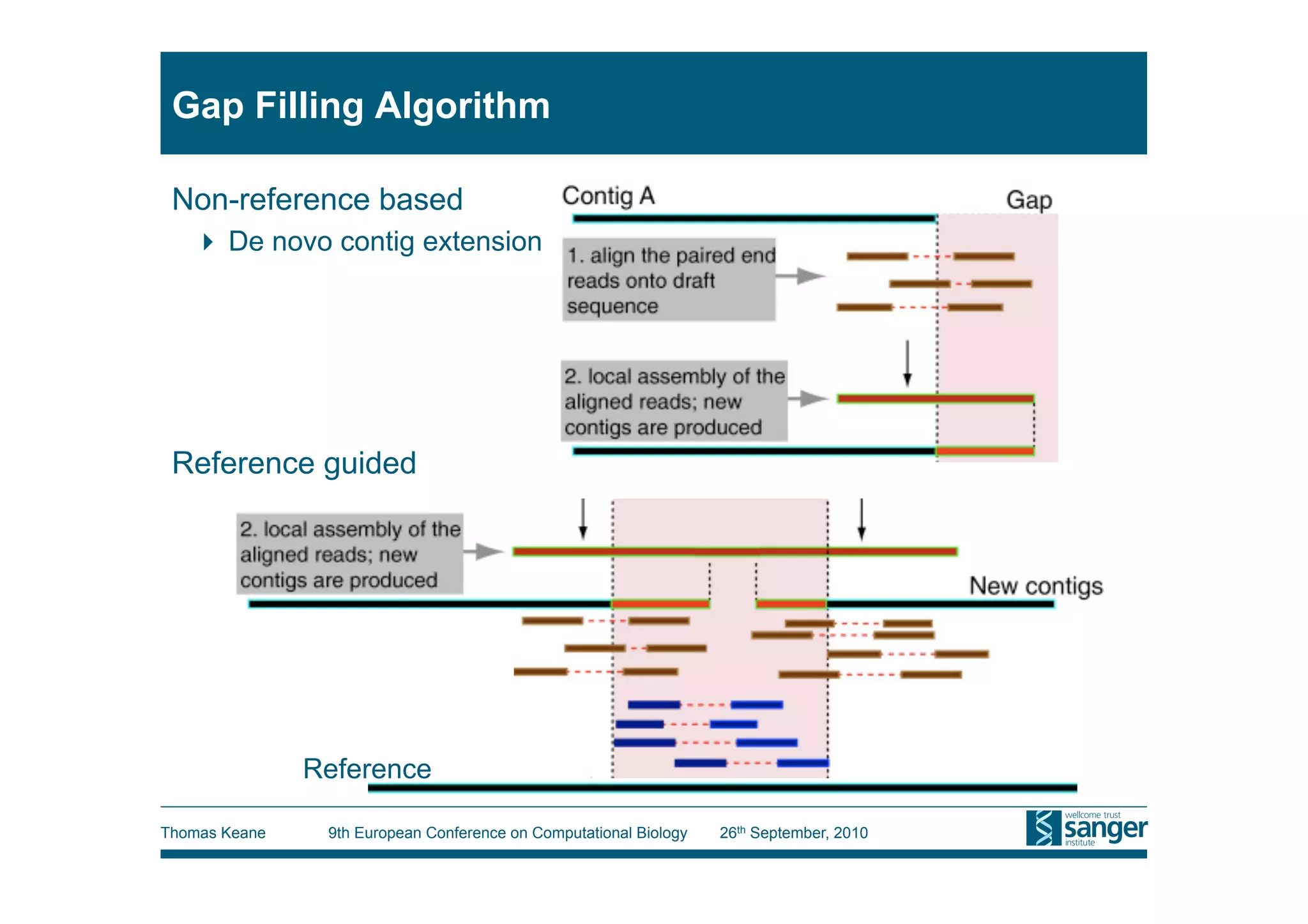

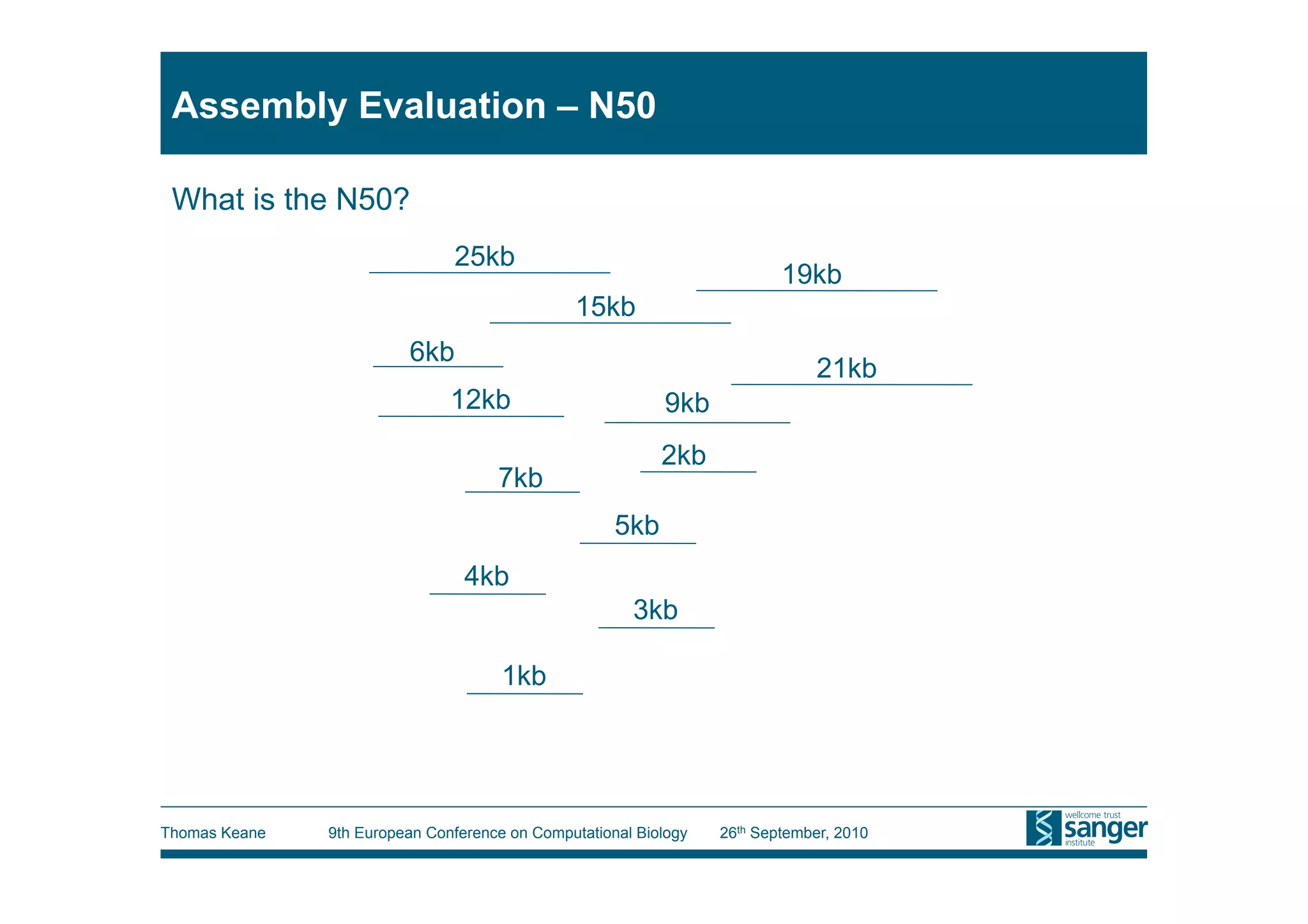


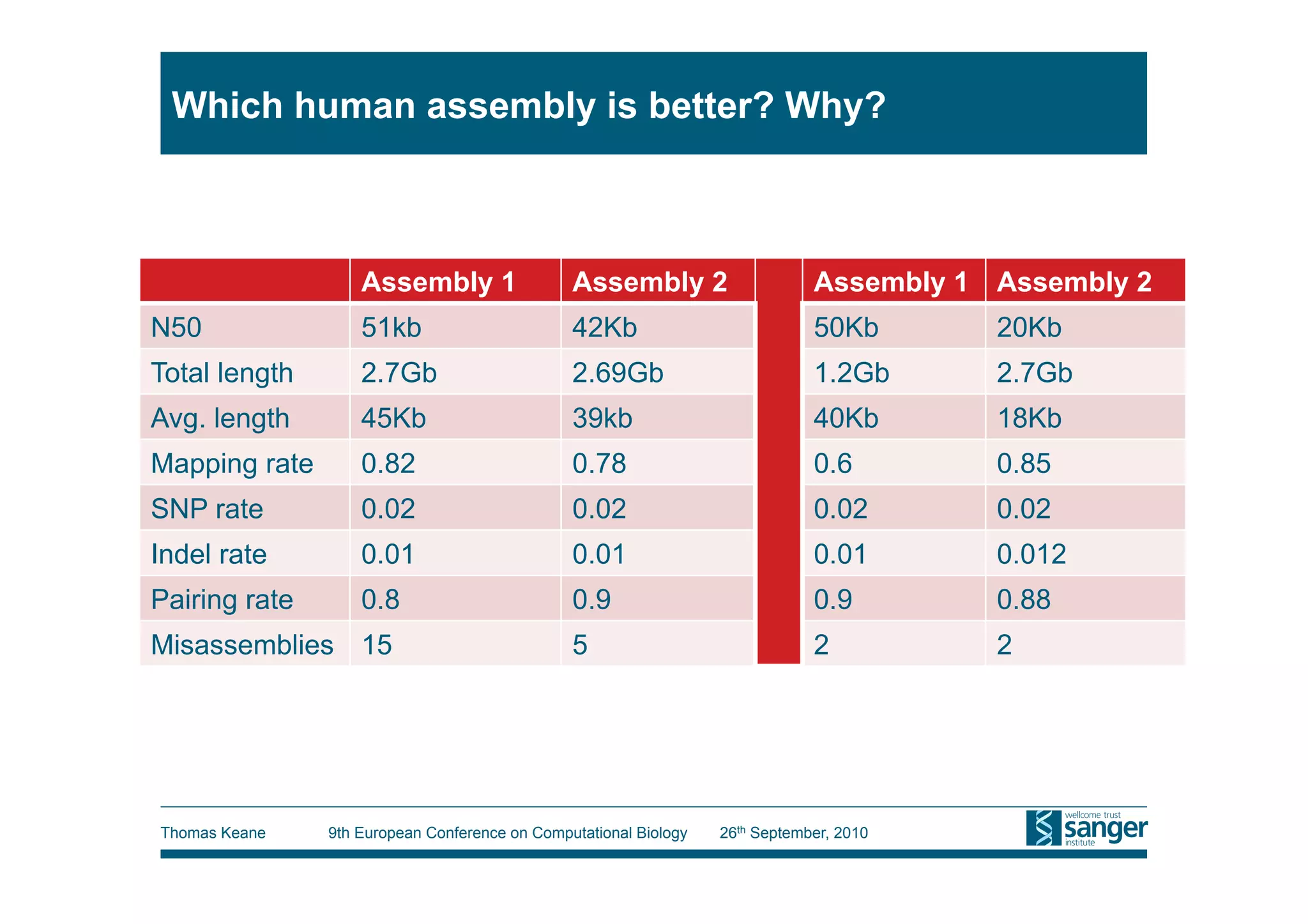





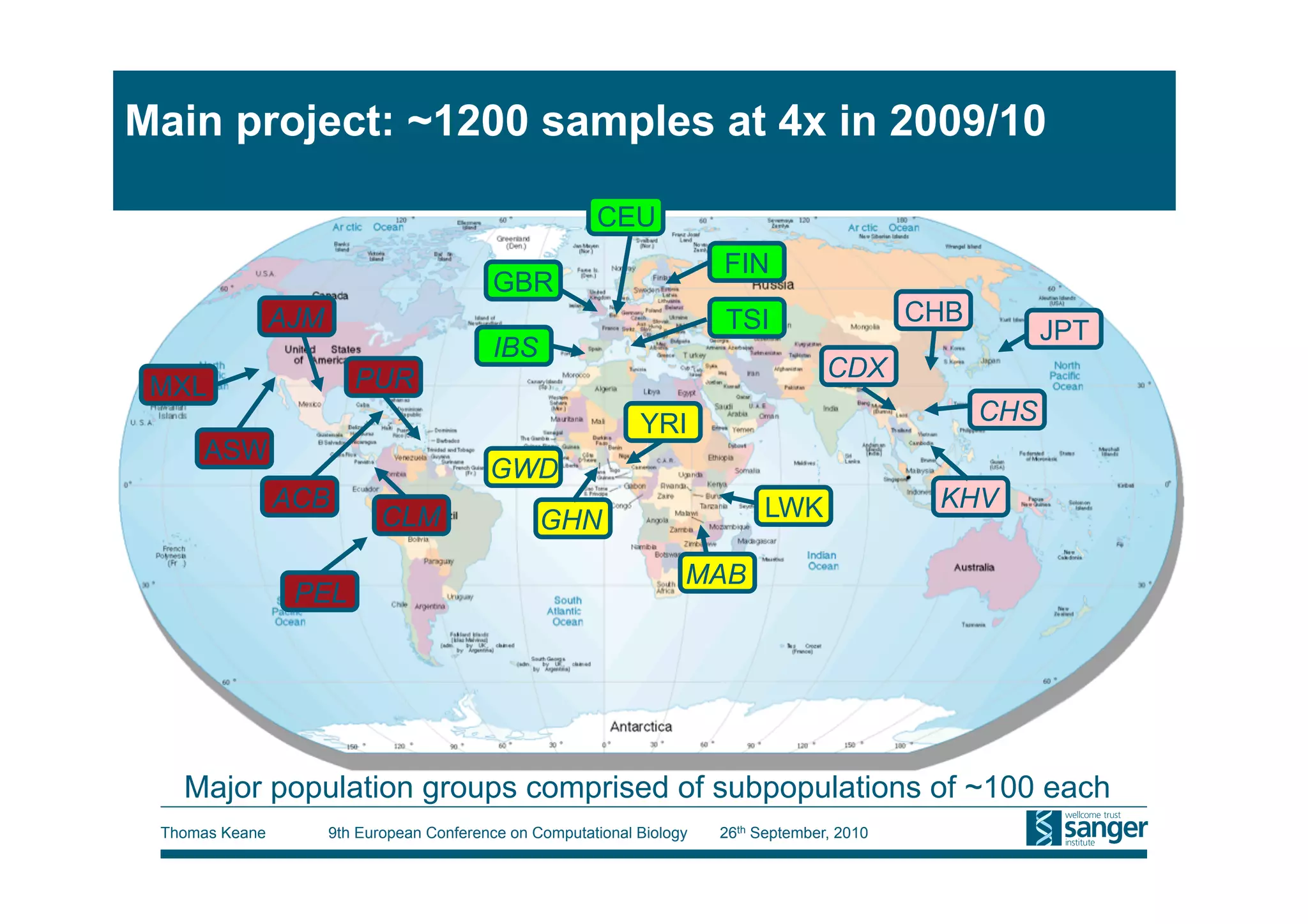
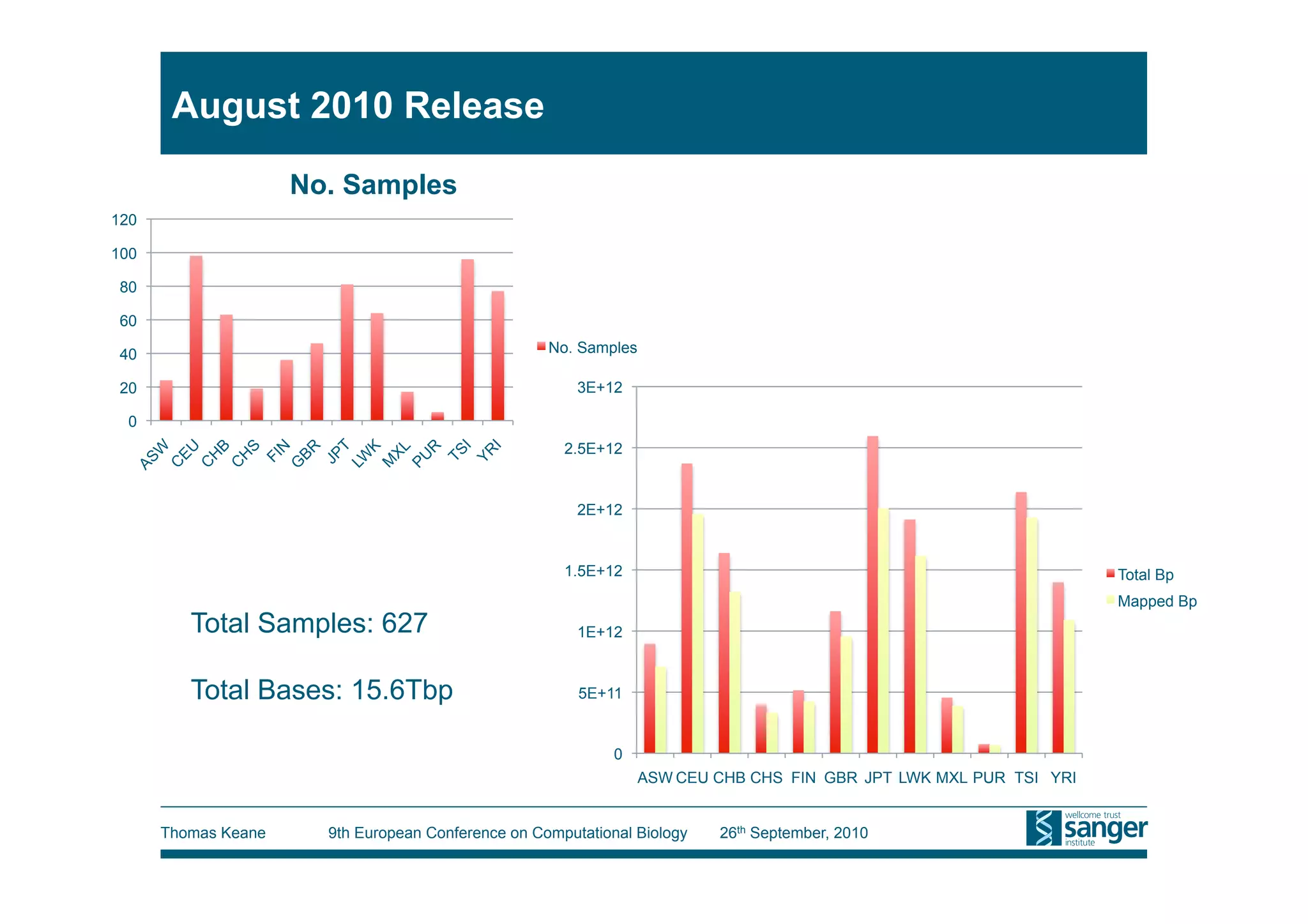
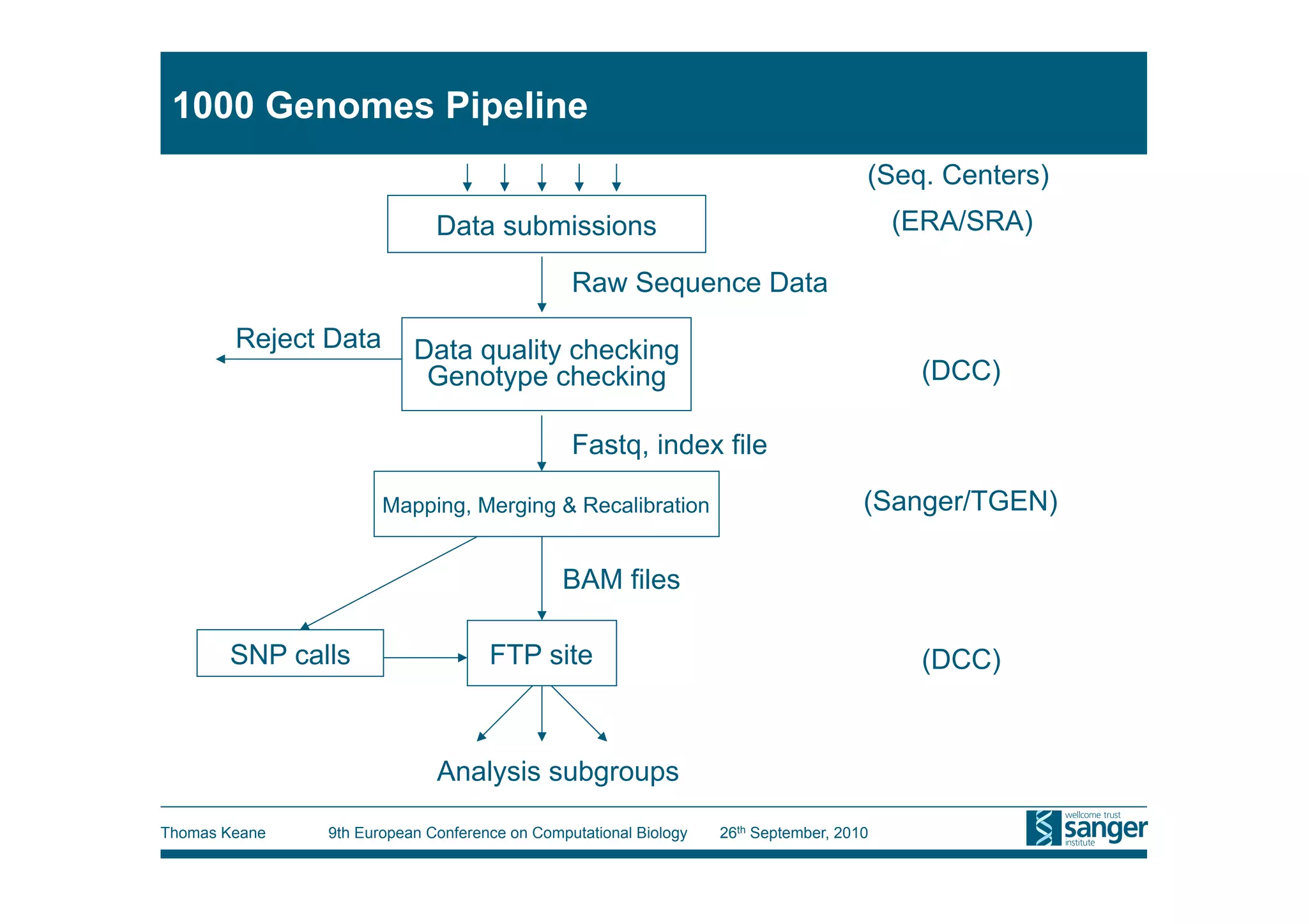
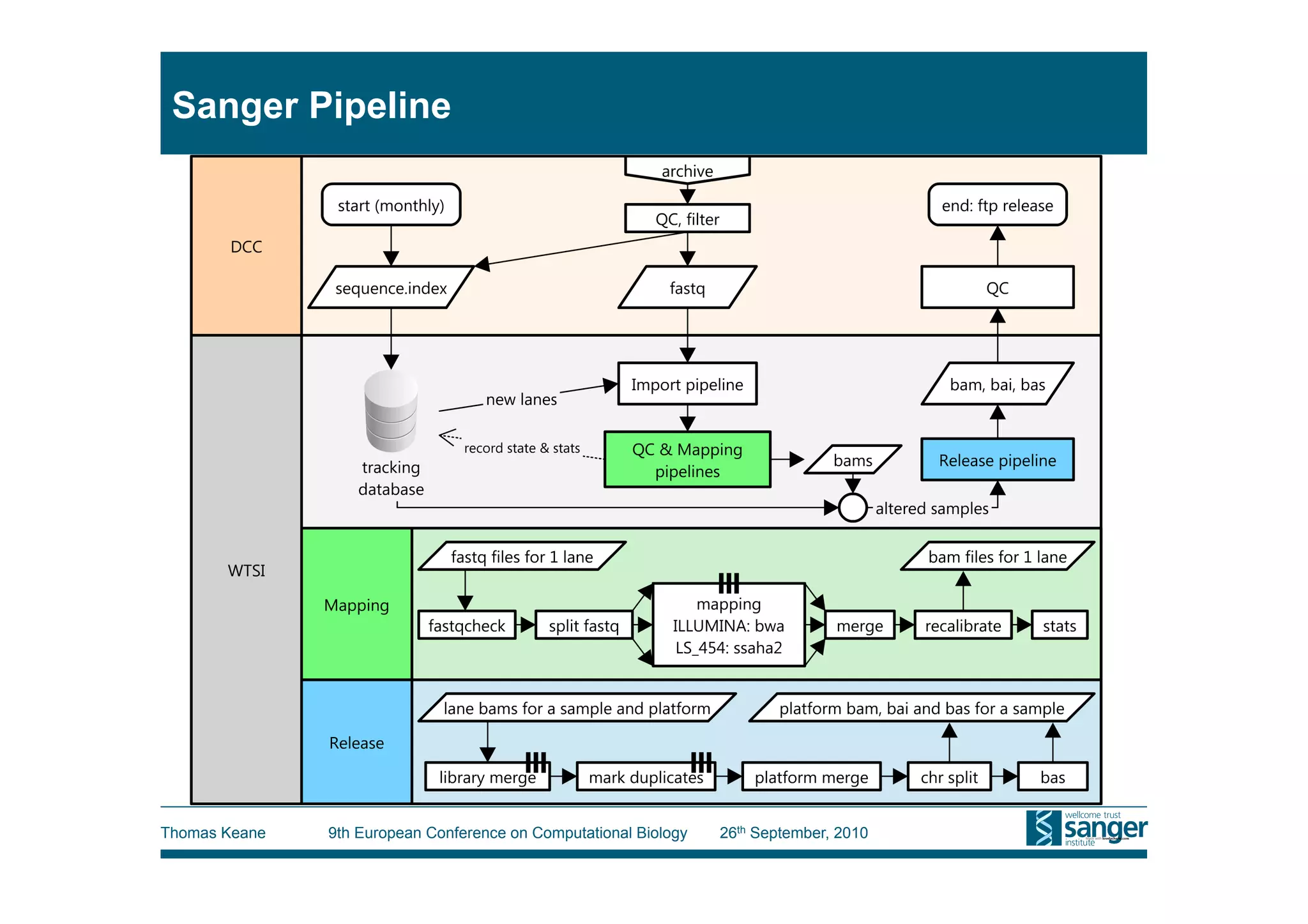
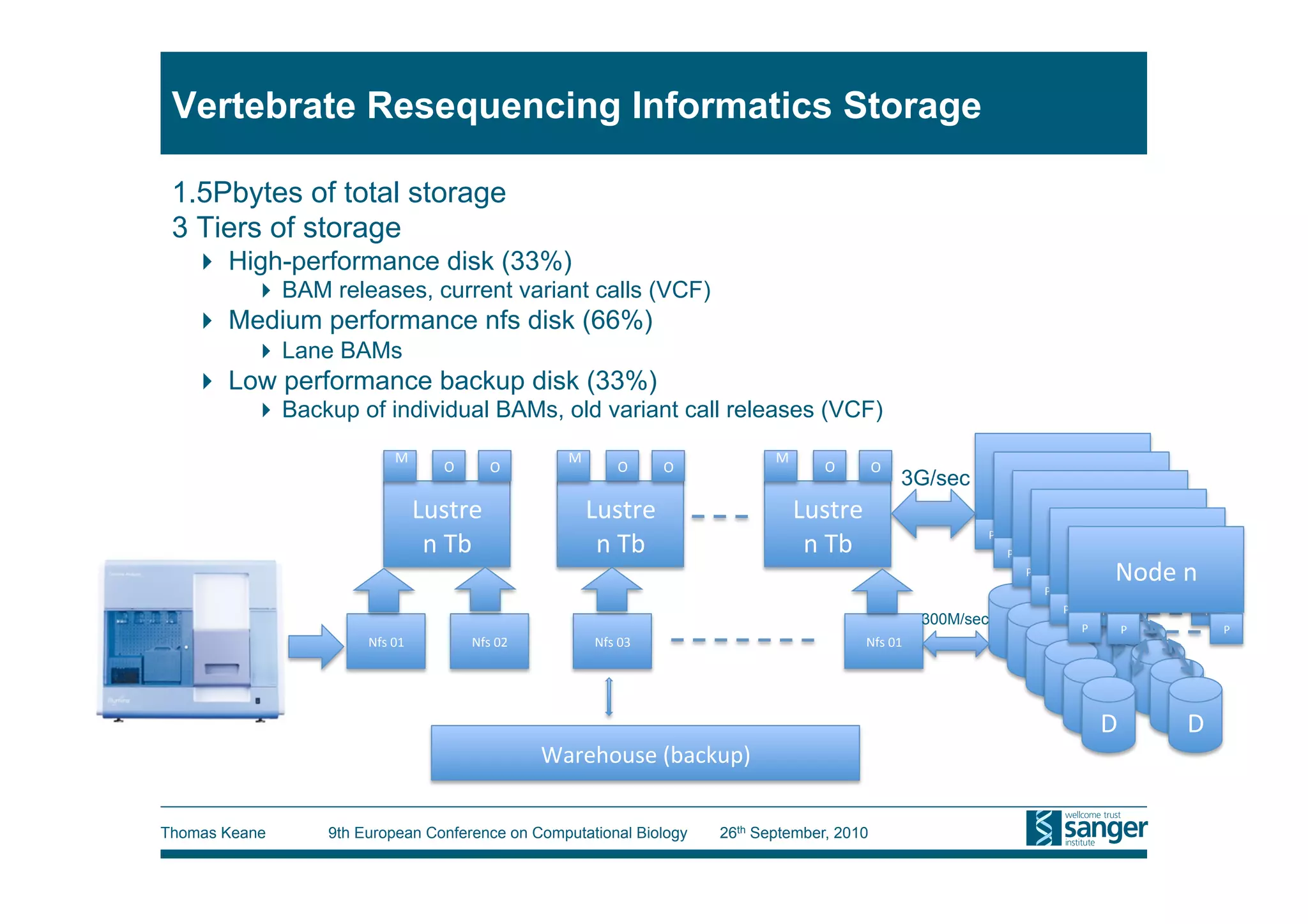






This tutorial provides an overview of working with next-generation sequencing data, including quality control, alignment, and variation analysis. It covers topics such as next-gen sequencing technologies and applications, quality control measures, short read alignment algorithms and tools, sequence assembly methods, and calling variants from sequencing data. The tutorial is presented by Thomas Keane and Jan Aerts at the 9th European Conference on Computational Biology.




































































































