Downloaded 125 times









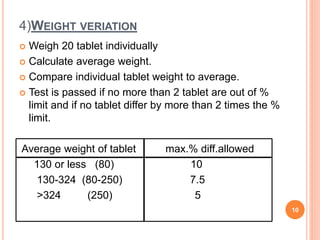


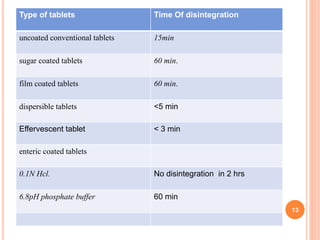

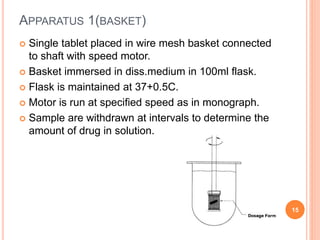
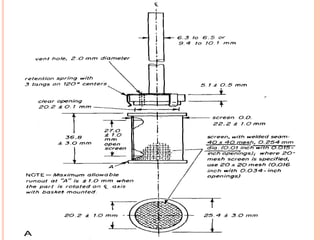


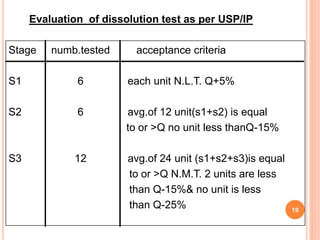








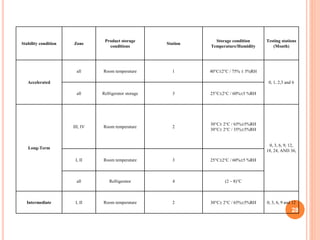











The document discusses evaluation and stability studies of tablets. It provides details on common tablet tests performed during evaluation including general appearance, hardness, friability, weight variation, disintegration, and dissolution. It also discusses factors affecting drug stability and the various types of stability that must be considered, including chemical, physical, microbiological, therapeutic, and toxicological stability. Guidelines for stability testing from ICH, USP, FDA and other organizations are also summarized regarding testing conditions, frequency, and requirements for re-testing tablets after registration.























































































