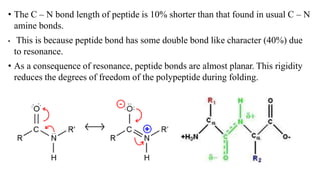
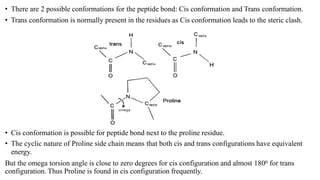
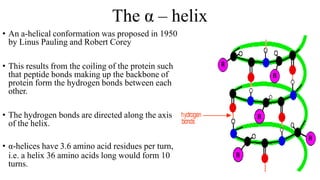
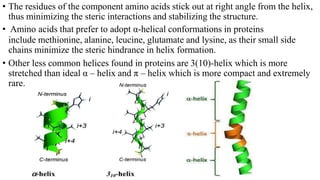
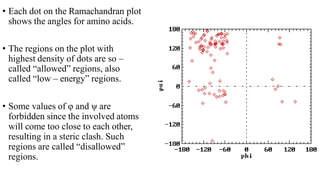
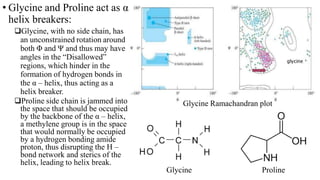
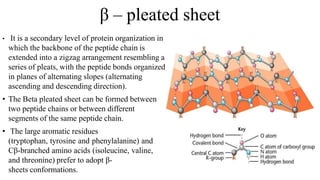
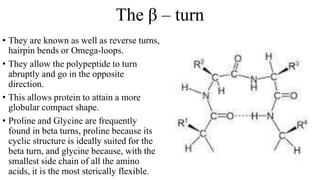
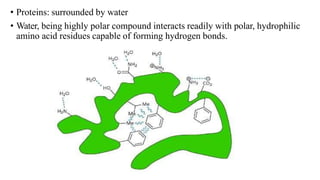
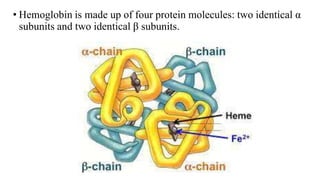
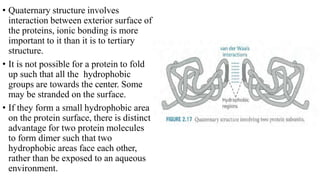
The document describes the four levels of protein structure:
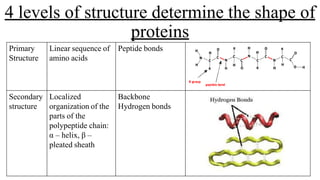
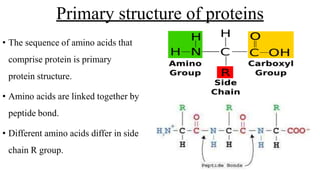
1) Primary structure is the linear sequence of amino acids joined by peptide bonds.
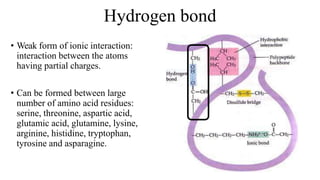
2) Secondary structure involves localized patterns like alpha helices and beta sheets formed by hydrogen bonds.
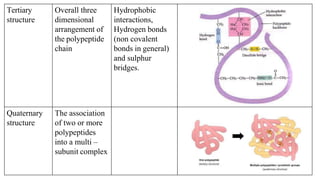
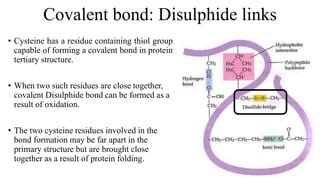
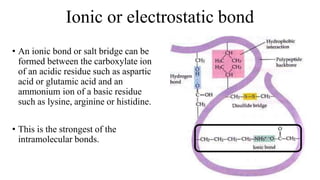
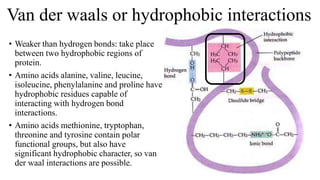
3) Tertiary structure is the overall 3D shape of the polypeptide chain formed by interactions like disulfide bridges, hydrogen bonds, and hydrophobic interactions.
4) Quaternary structure refers to complexes of multiple polypeptide subunits.