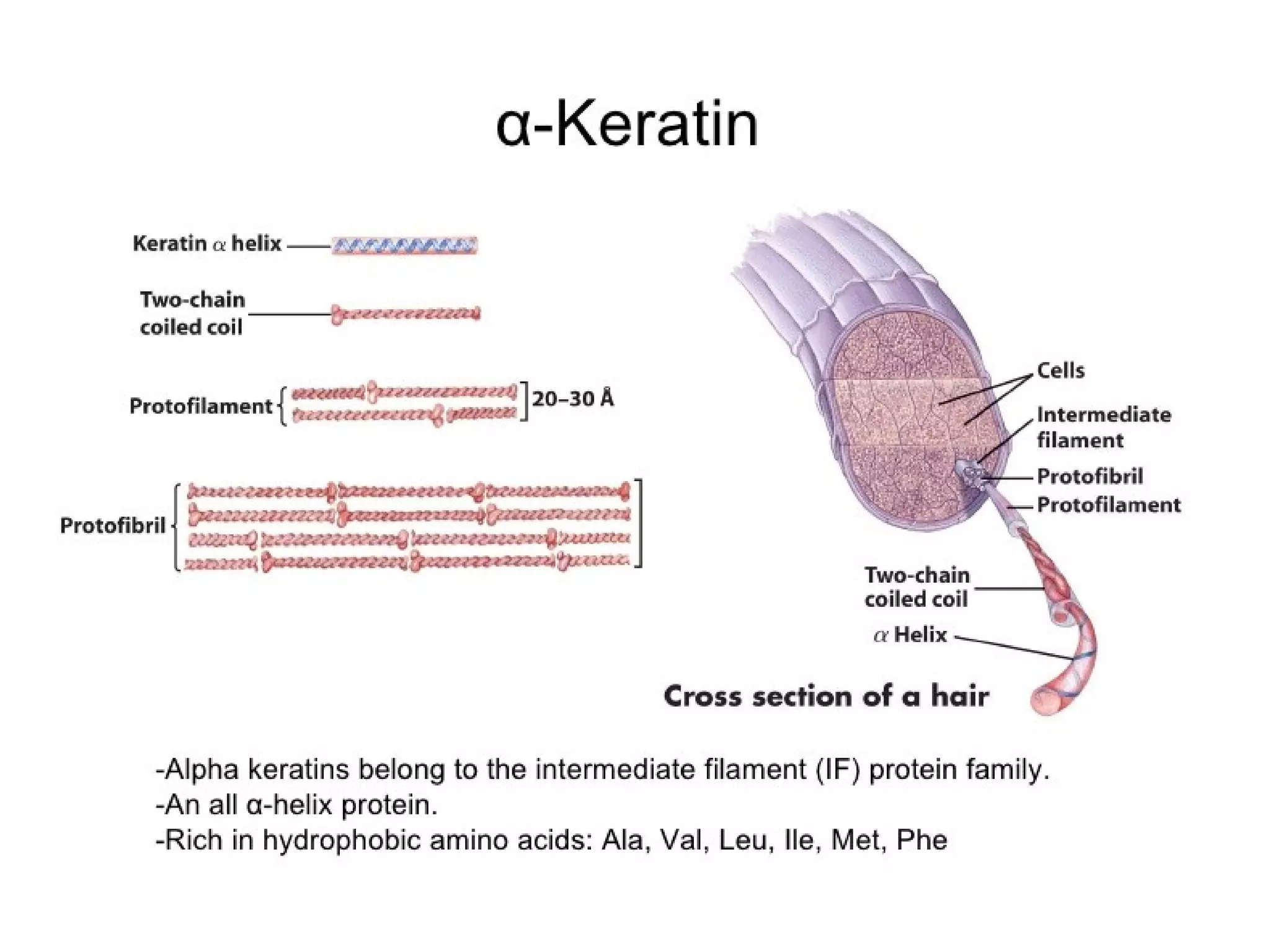
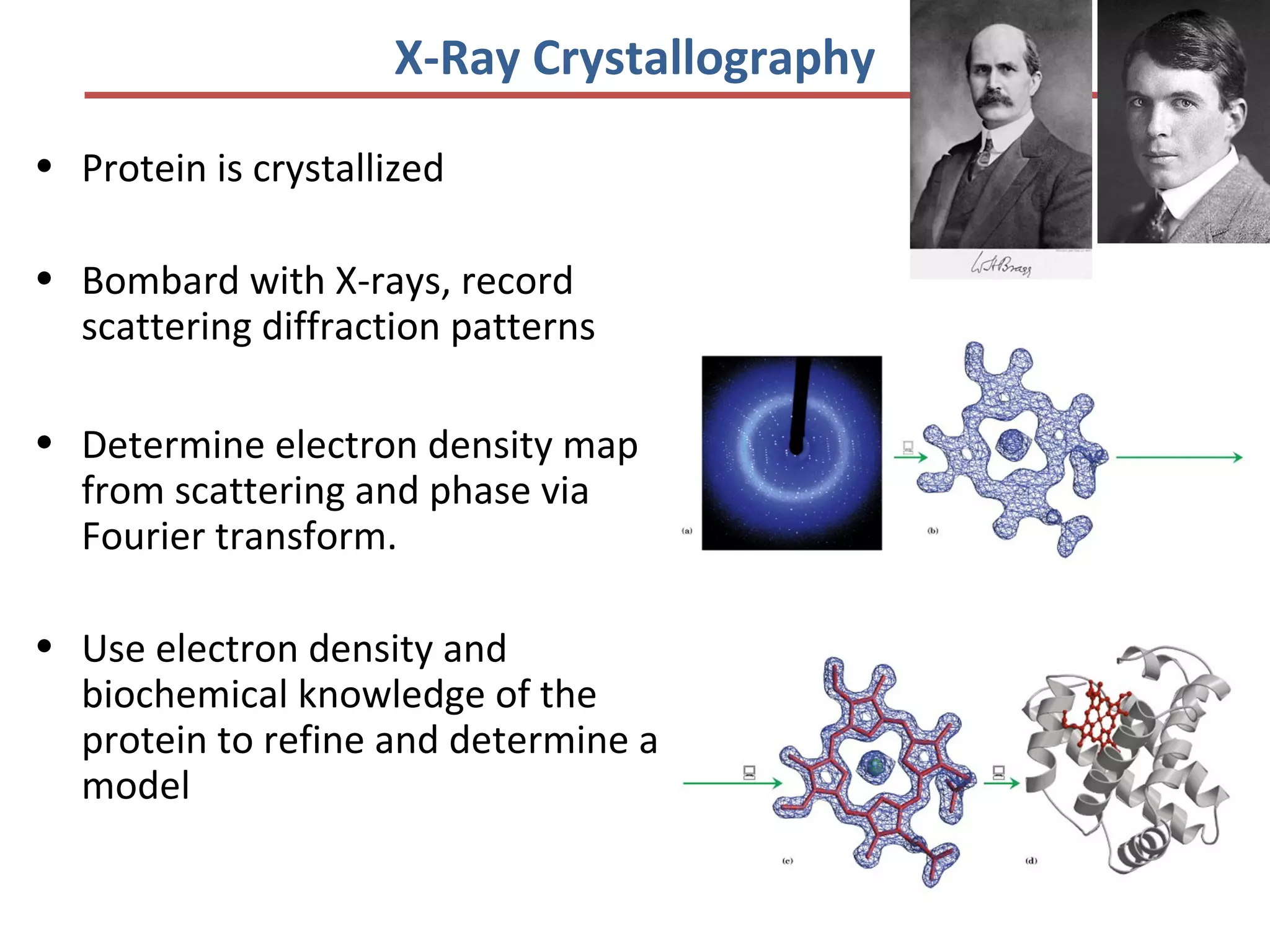
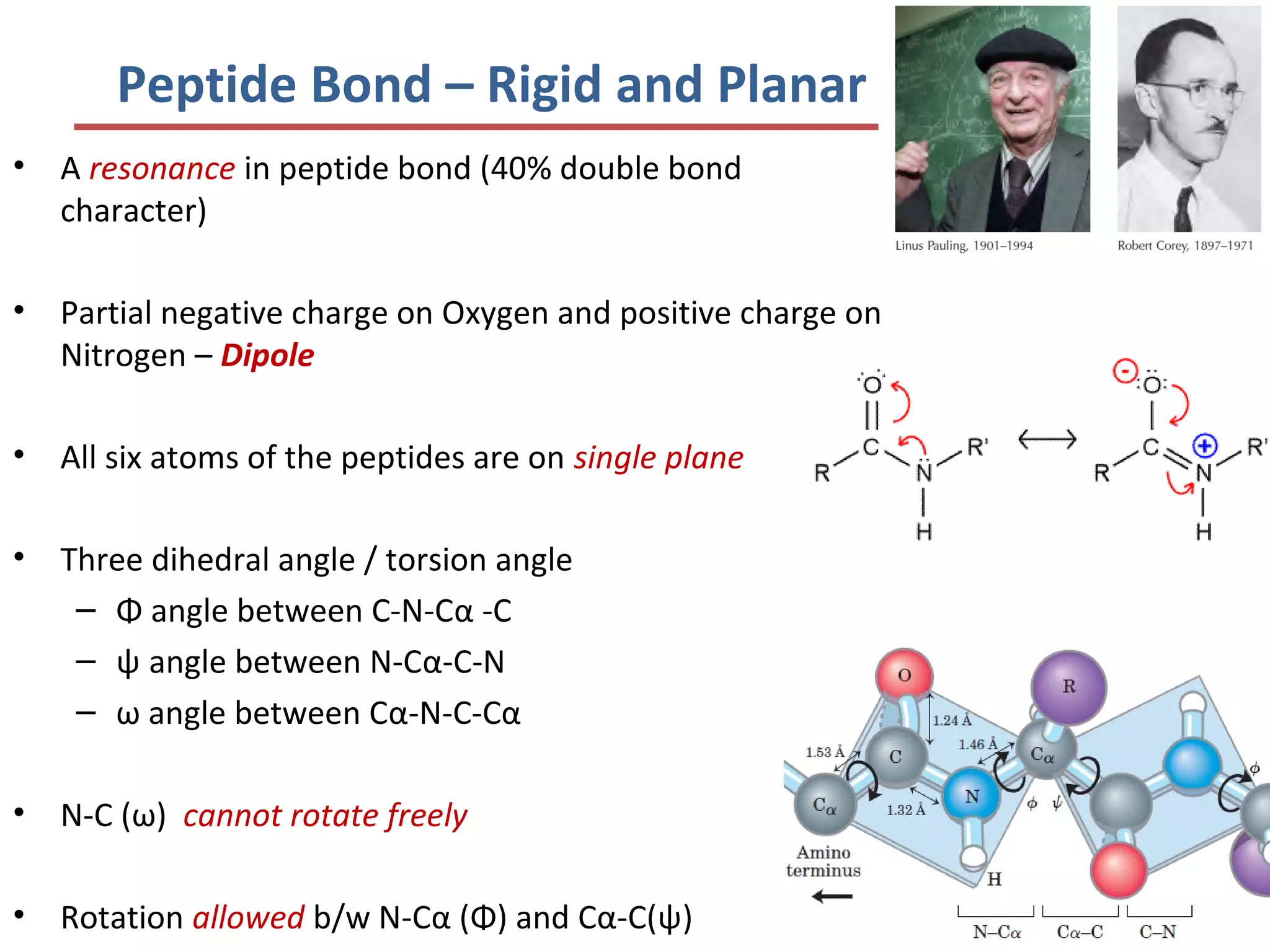
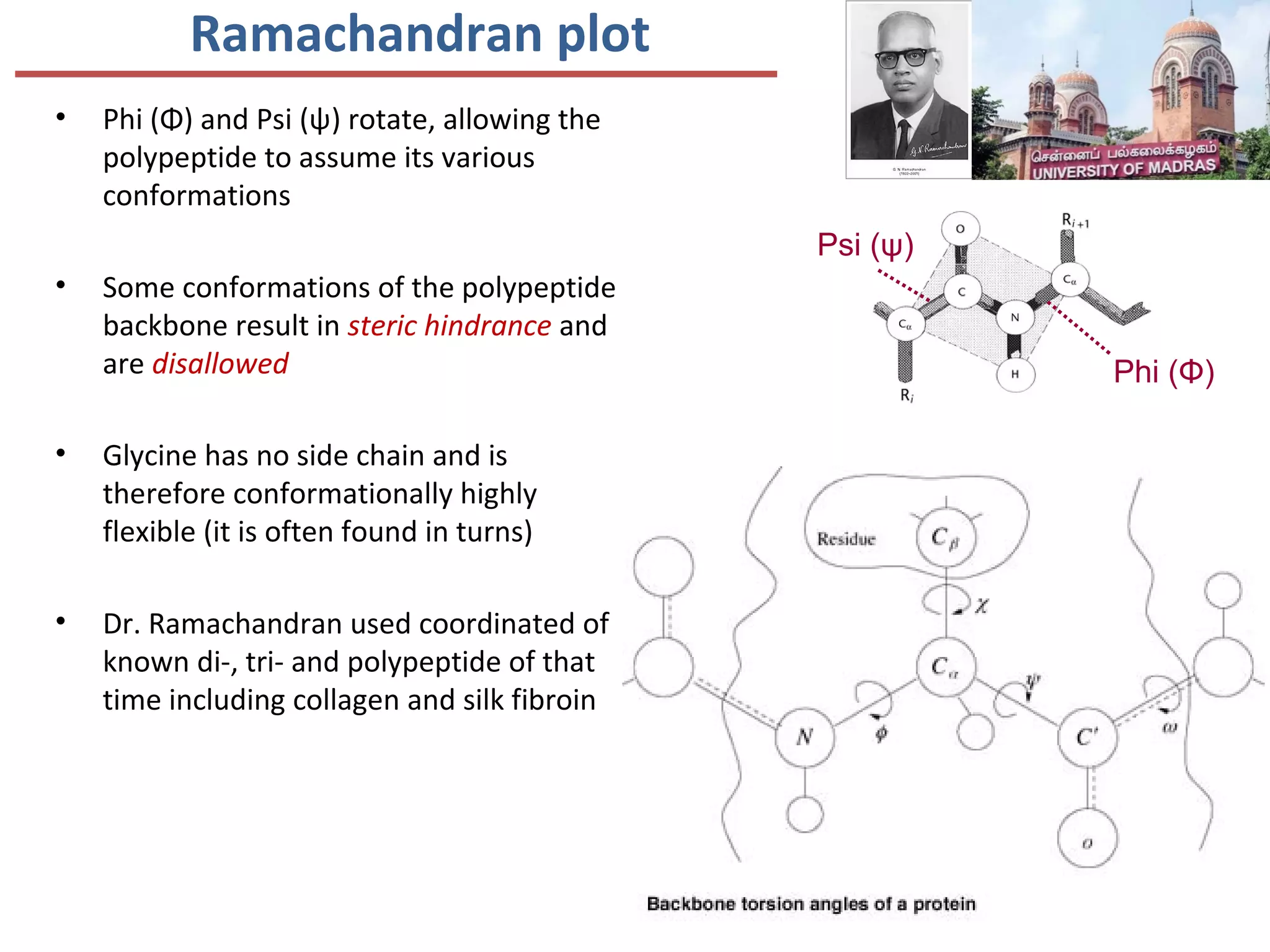
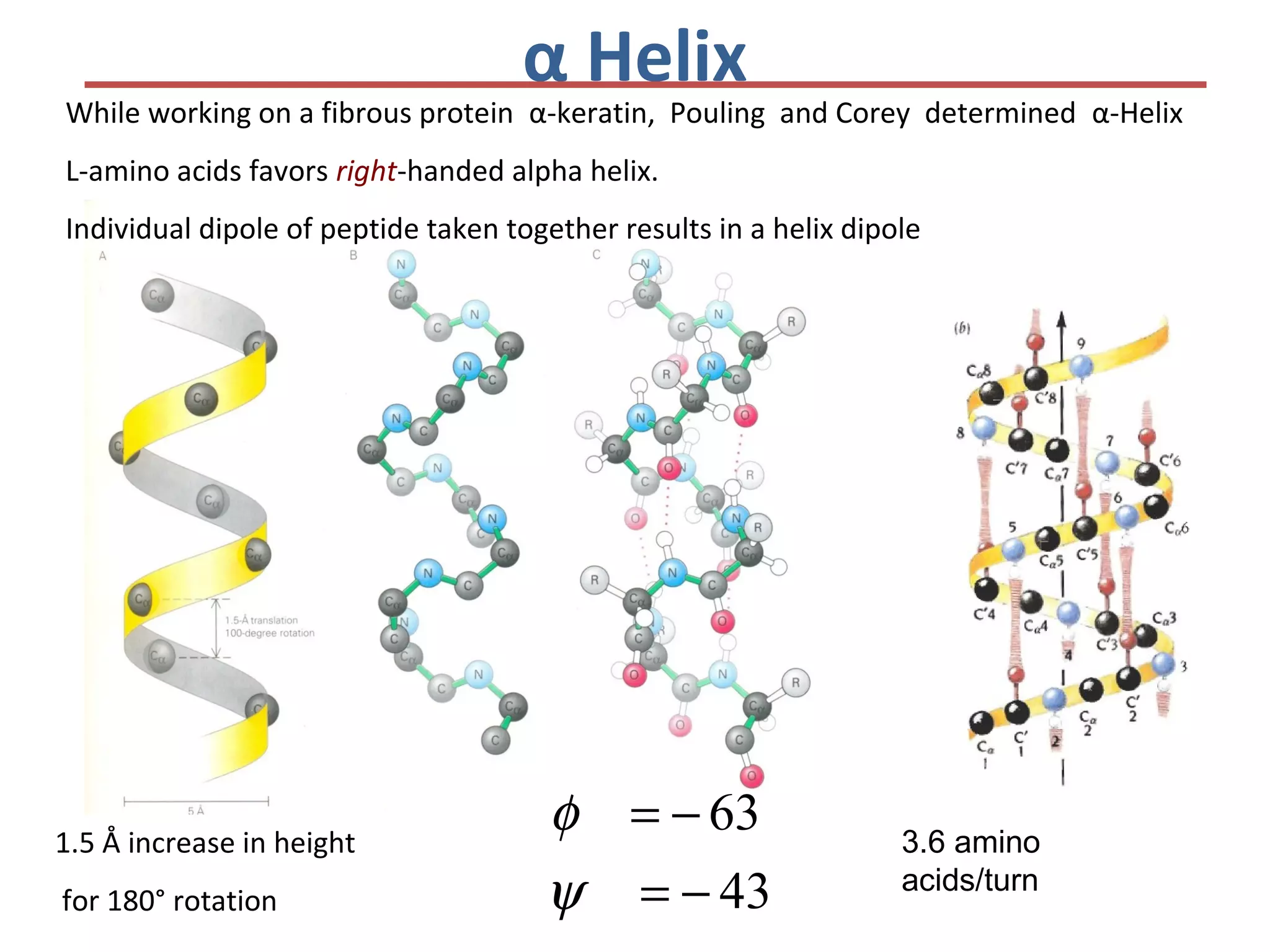
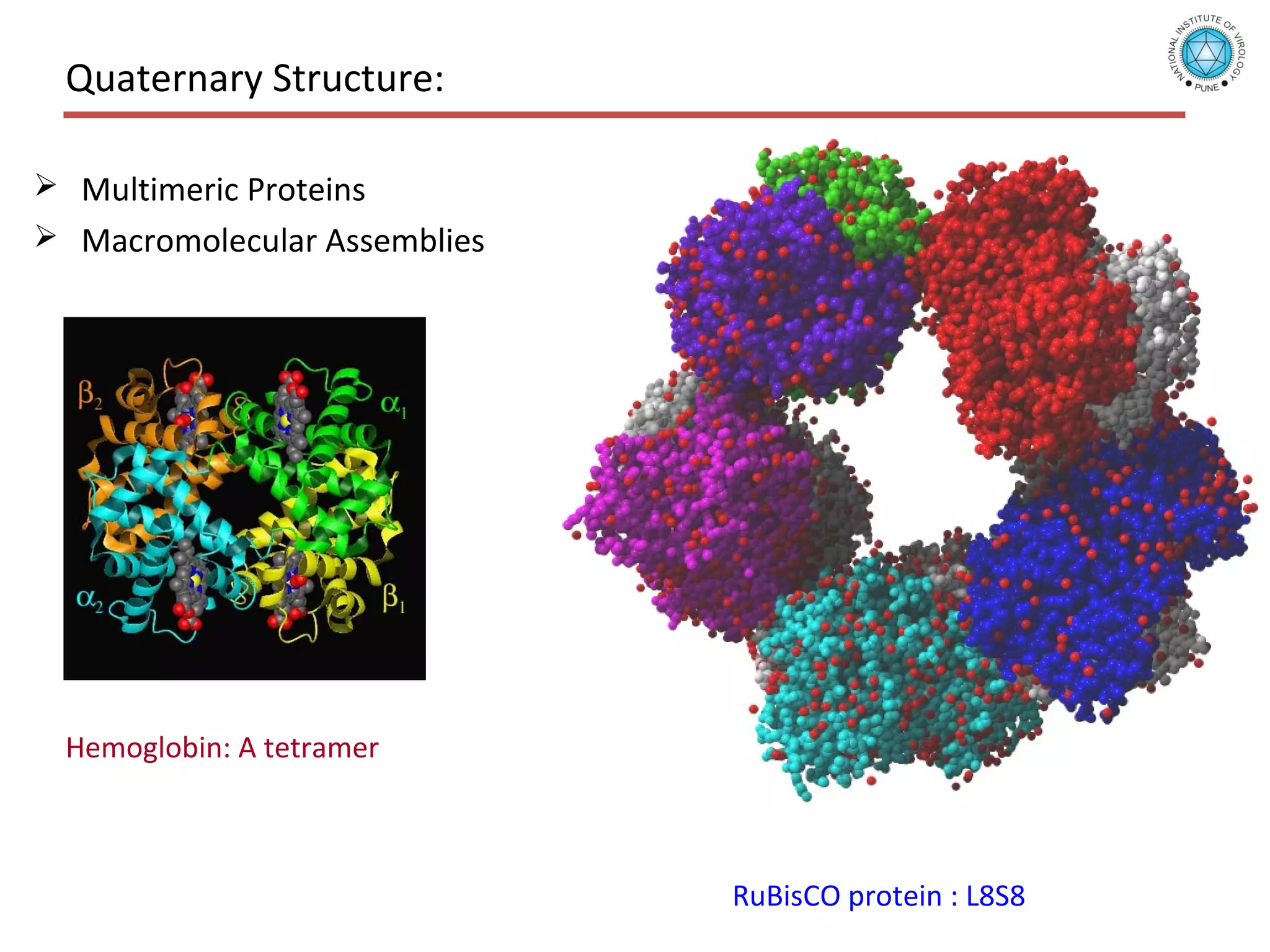
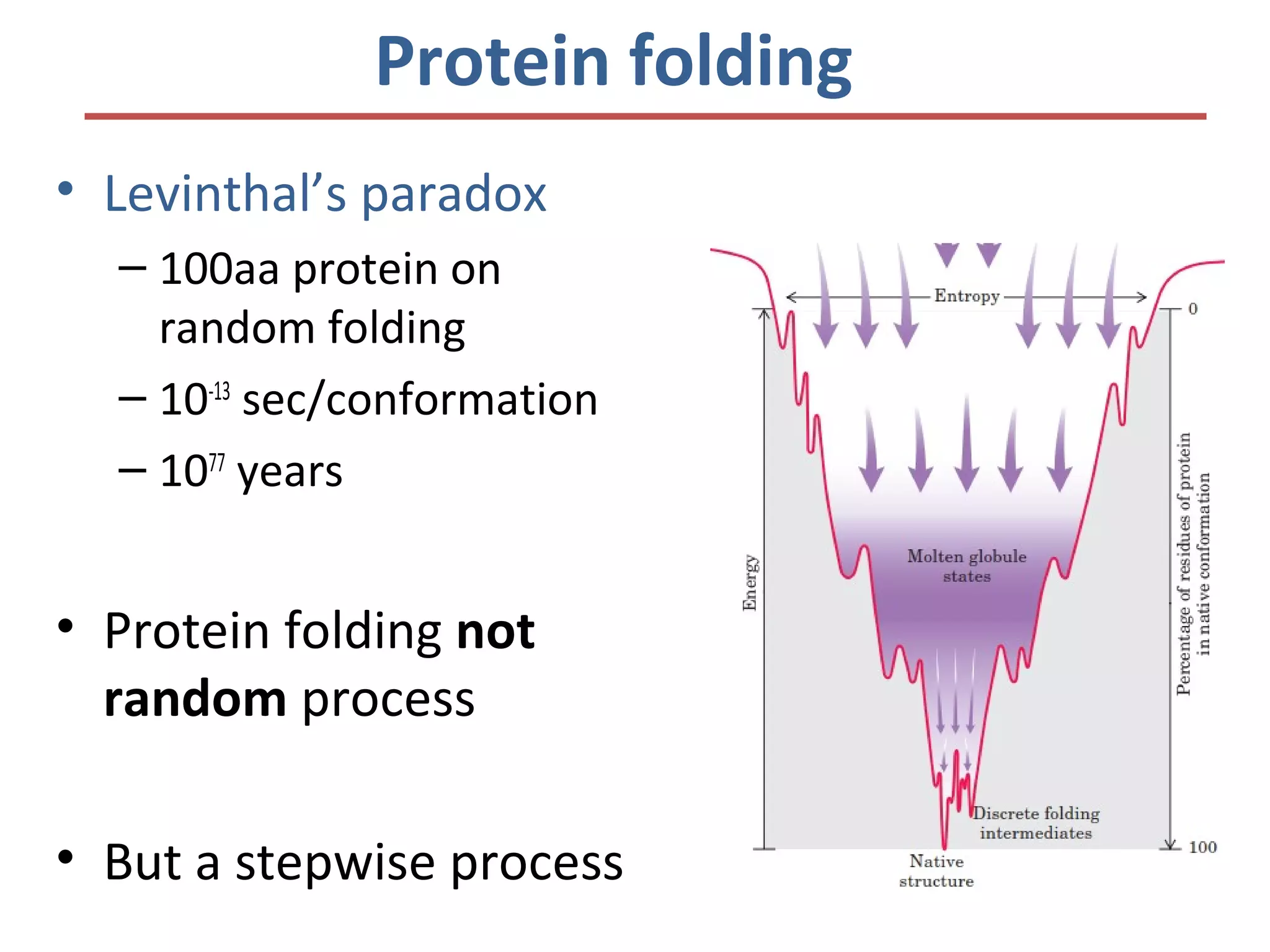
This document discusses the structure and function of various proteins, including fibrous proteins like keratin and collagen, their hierarchical structures, and the principles governing protein conformation. It explains how proteins fold into their functional shapes through interactions such as hydrogen bonding, and methods for determining protein structures, including x-ray crystallography. The document also touches upon intrinsically disordered proteins, protein denaturation, and the implications of specific mutations like in sickle-cell anemia.