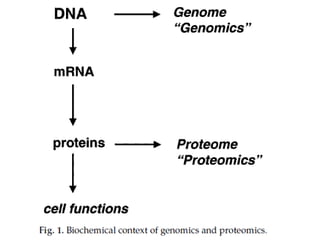
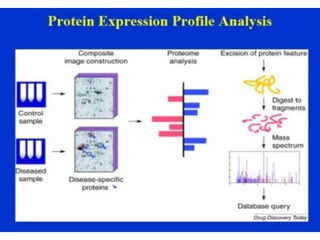
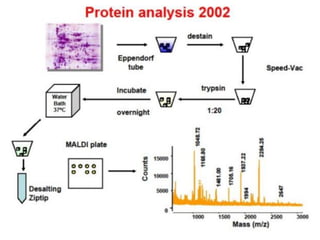
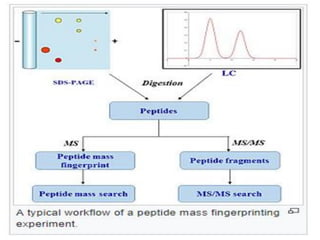
Proteomics is the study of the entire set of proteins produced by a cell type, examining how proteins interact with cellular processes and the environment. It aims to highlight differences in proteomes under various conditions, with applications in cancer research for early detection and individualized treatment plans. Key techniques in proteomics include mass spectrometry, x-ray crystallography, nuclear magnetic resonance, and protein microarrays to analyze protein structure and functionality.



































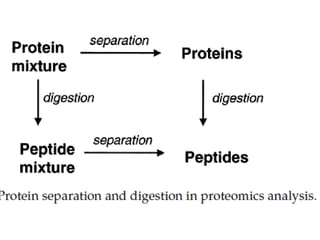


![• For proteomic analysis, the objective here is to recover
as much of the protein as possible with as little
contamination by other biomaterials (e.g., lipids,
cellulose, nucleic acid, etc.) as possible.
• This is generally done with the aid of:
• Detergents (e.g., SDS, 3-([3-cholamidopropyl] dimethyl
ammonio)-1- propane sulfonate (CHAPS), cholate, Tween),
which help to solubilize membrane proteins and aid their
separation from lipids
• Reductants (e.g., dithiothreitol [DTT], mercaptoethanol,
thiourea), which reduce disulfide bonds or prevent protein
oxidation](https://image.slidesharecdn.com/proteomicstechniquesapplications-220828035128-7b52109c/85/Proteomics-techniques-applications-pdf-41-320.jpg)





































































































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)
