Downloaded 574 times










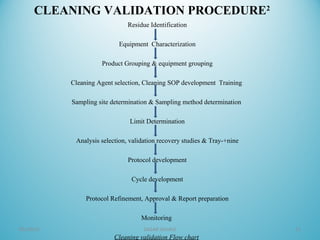
























This document outlines the importance and procedures of cleaning validation in the pharmaceutical industry, highlighting its necessity for ensuring product safety and compliance with regulatory requirements. It details the processes involved in establishing acceptance criteria, analytical methods, and the challenges faced in cleaning validation. The conclusion emphasizes the critical role of cleaning validation in preventing contamination during drug manufacturing to maintain high quality and safety standards.











































































































