Downloaded 250 times







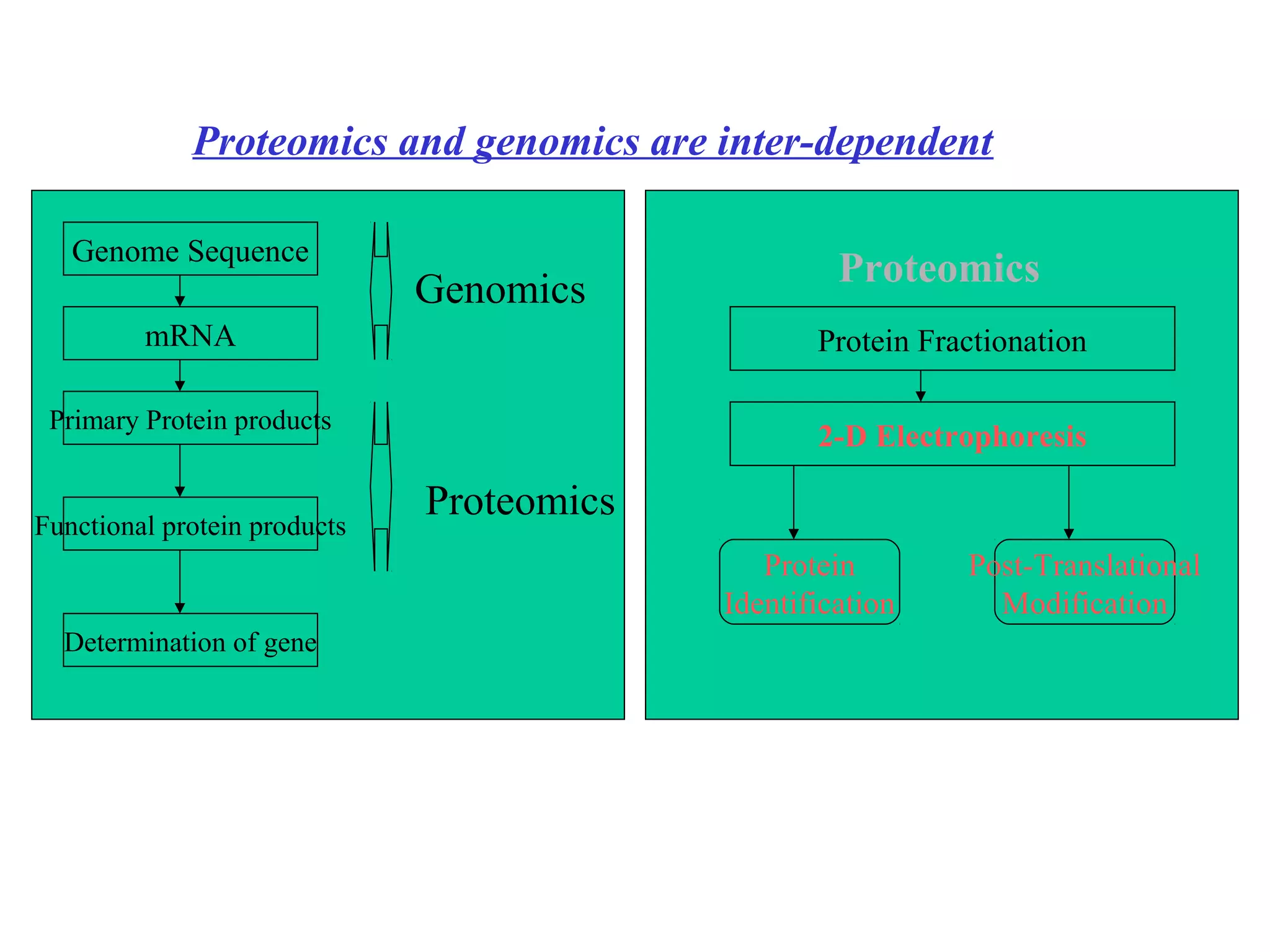



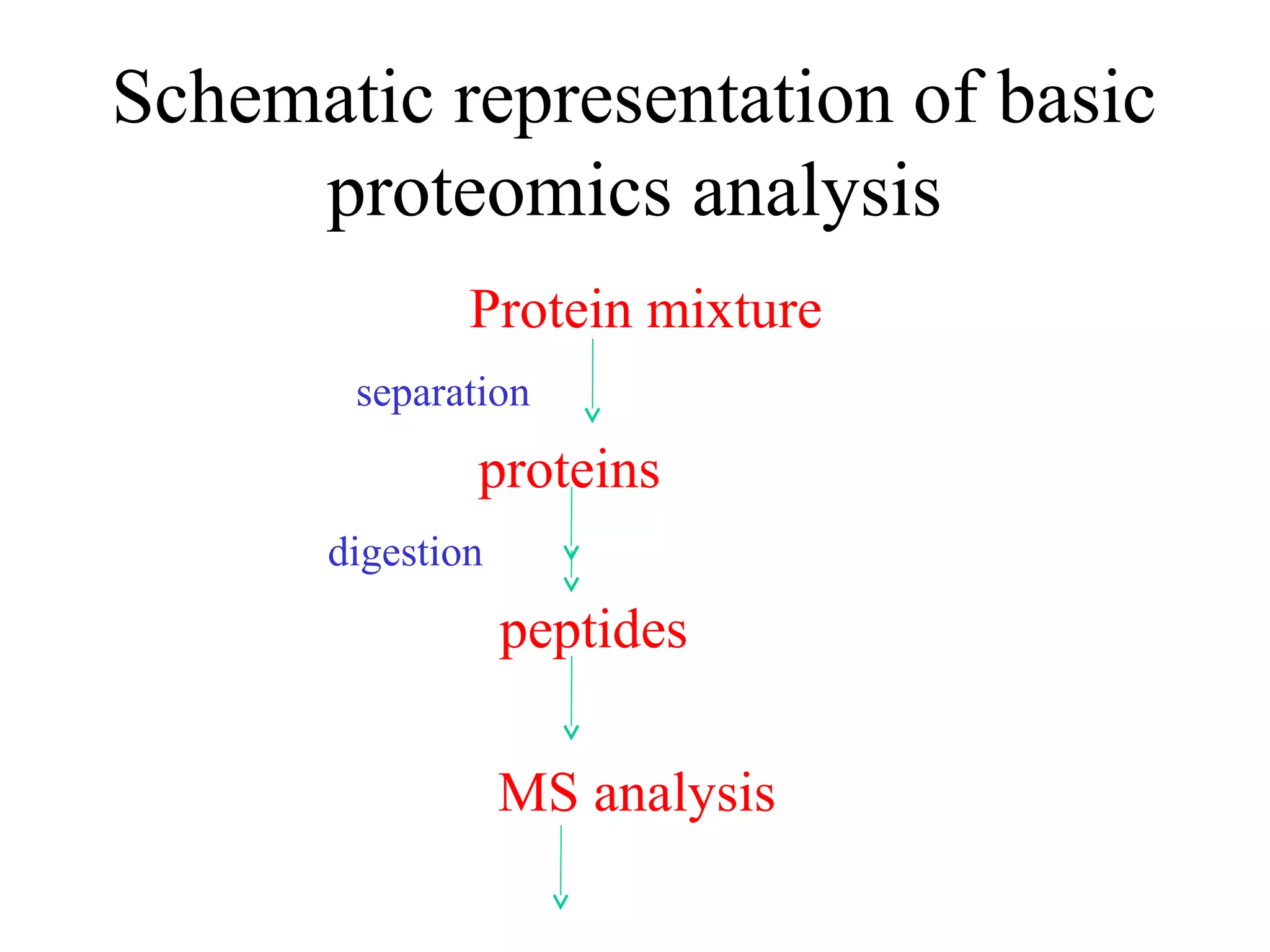







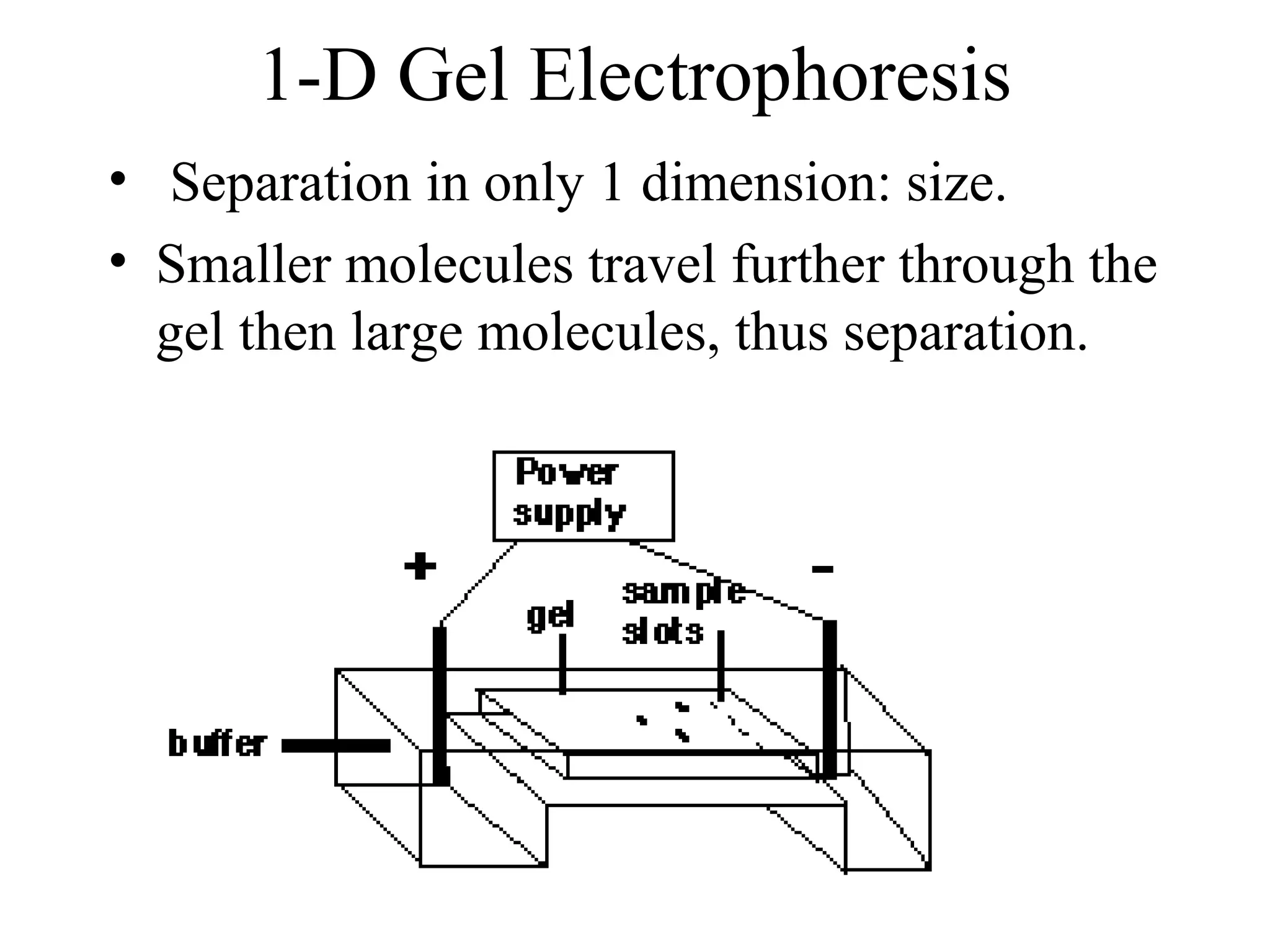



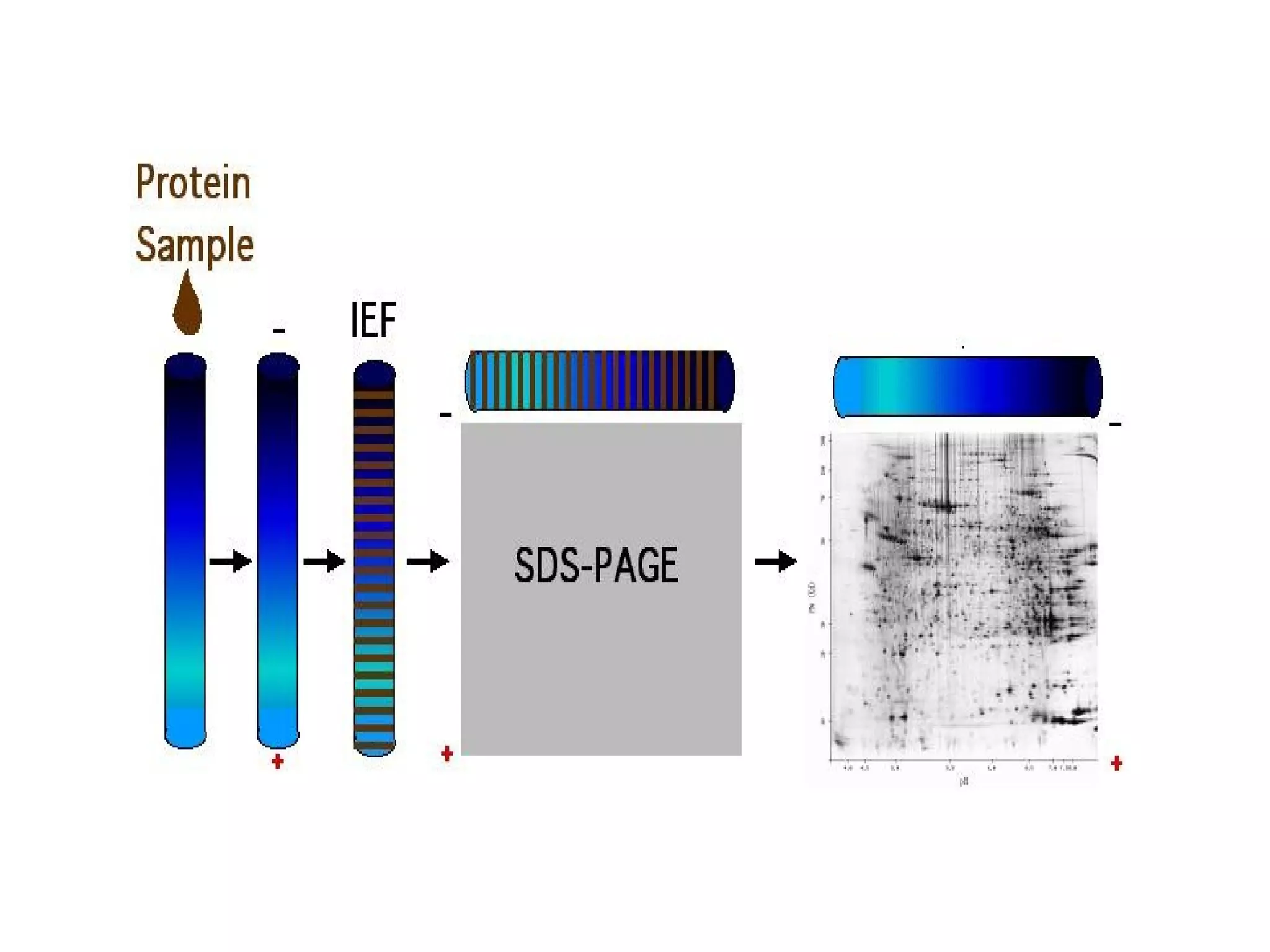








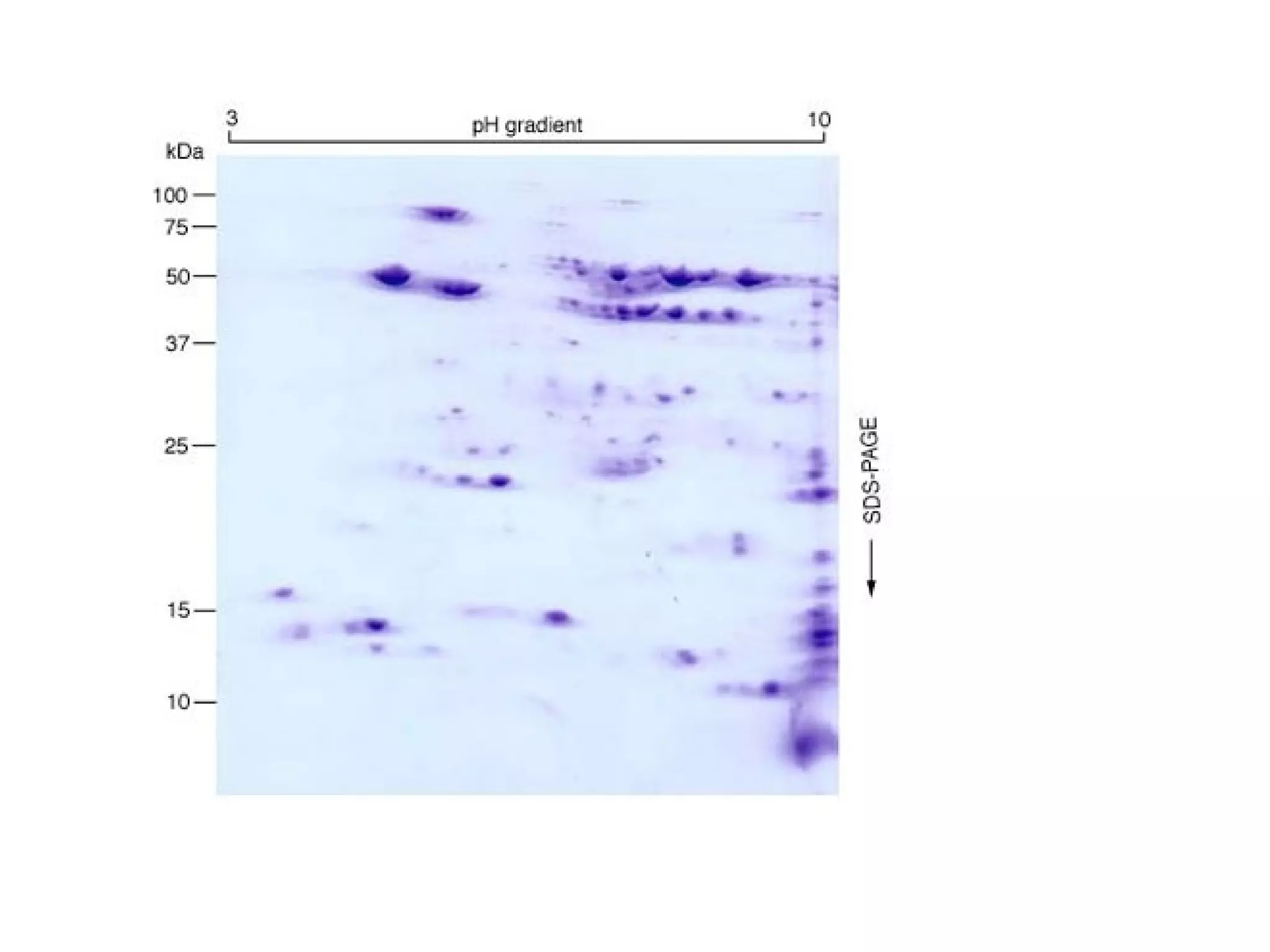















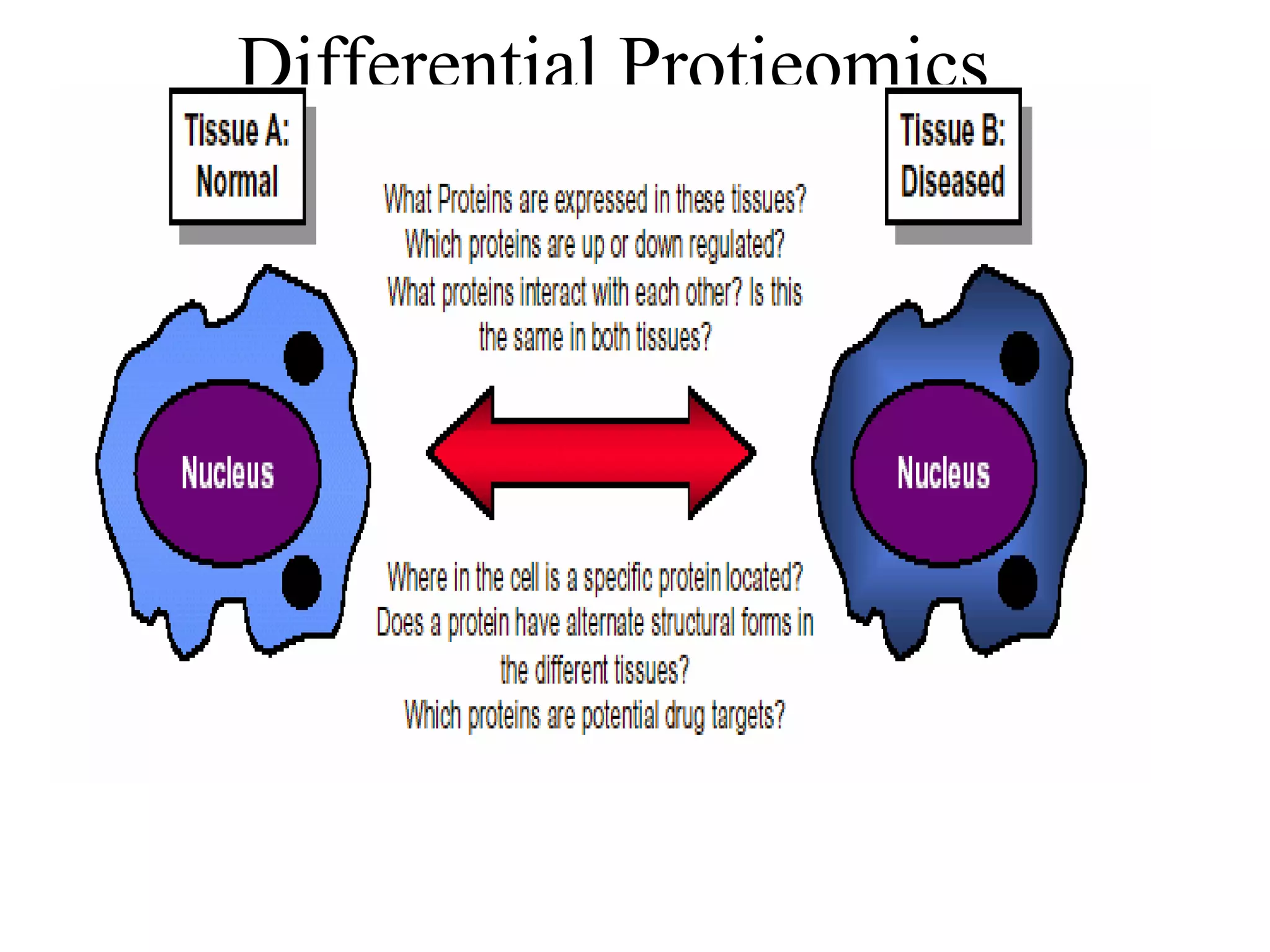
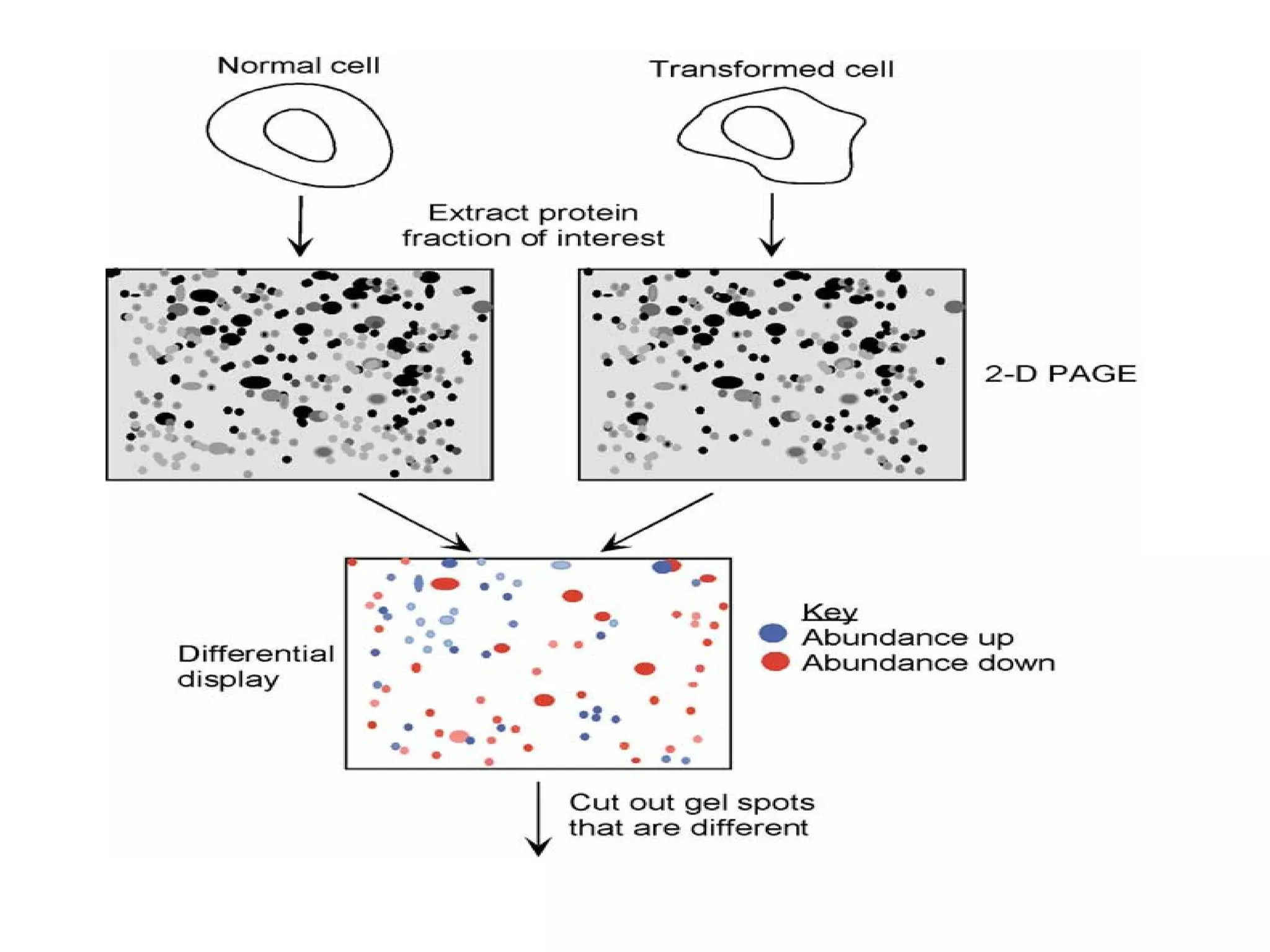

The document discusses proteomics, which is the study of the entire complement of proteins in a cell or organism. It defines key proteomics terms like proteome and describes techniques used in proteomics like protein separation, 2D gel electrophoresis, mass spectrometry, and protein digestion. The goals of proteomics include detecting and comparing protein expression profiles to understand biological processes and discover drug targets. Proteomics provides important insights not available through genomics alone.































































