Download to read offline

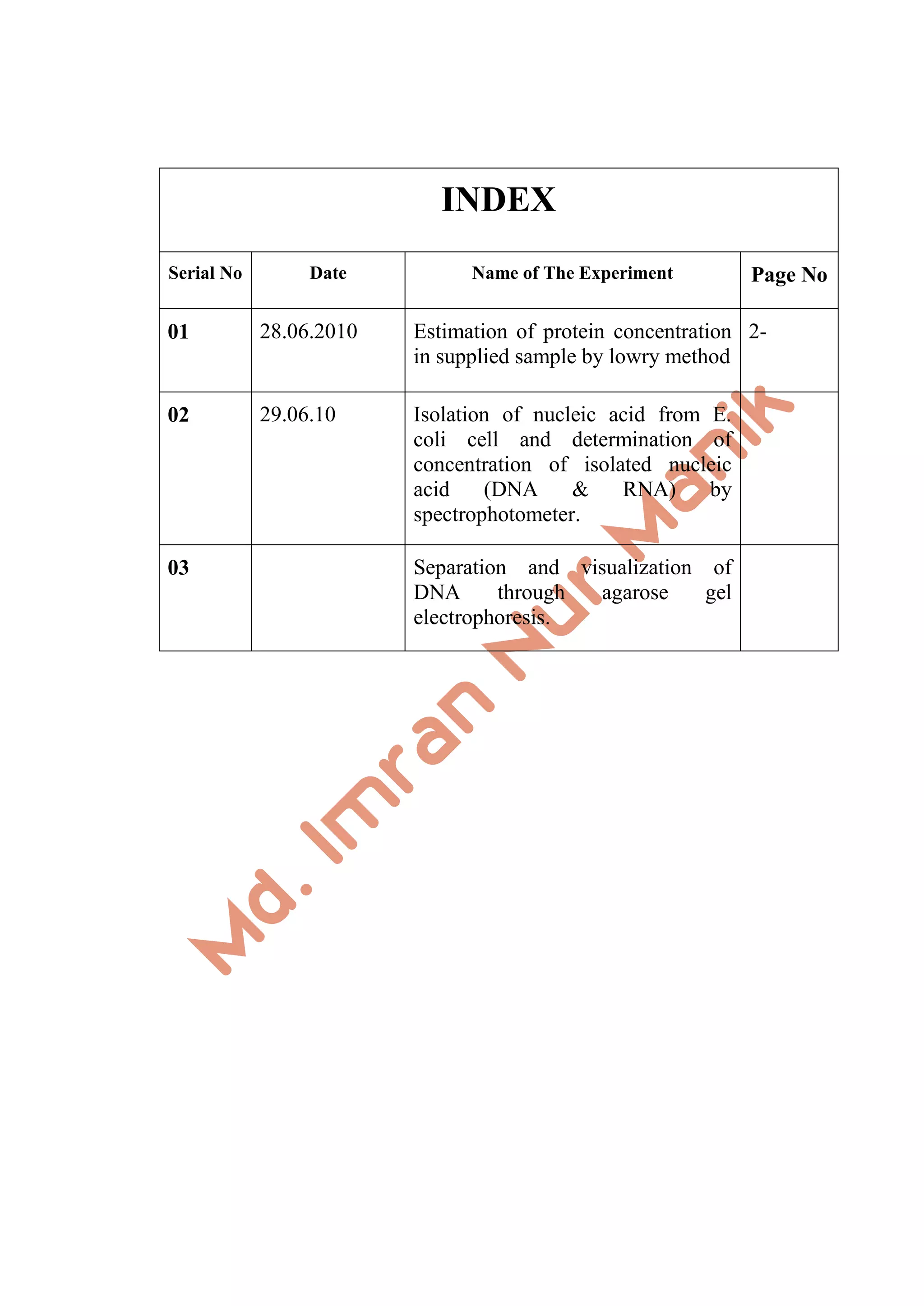




















This document provides instructions for performing three experiments: 1) Estimating protein concentration using the Lowry method. 2) Isolating nucleic acids from E. coli and measuring concentration using a spectrophotometer. 3) Separating and visualizing DNA using agarose gel electrophoresis. Reagents, equipment, and step-by-step procedures are described for each experiment. Calculations are shown for determining protein concentration from absorbance measurements.























































































