Downloaded 11 times







![Isolated microcephaly
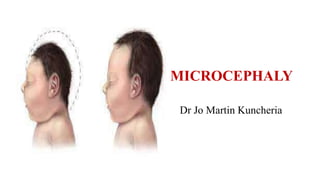
•Also called microcephaly vera, primary microcephaly or true microcephaly. Characteristic clinical
features of this phenotype include:
• Group of conditions which-
Do no usually have associated malformations
Follow mendelian pattern of inheritance or have association with specific genetic syndrome
• Infants are usually identified at birth
•Is present at birth and uncomplicated by anomalies outside the brain
•The brain may have normal architecture but is small (more than 3 standard deviations [SD] below the
mean)
• The more common types include familial and autosomal dominant microcephaly and a series of
chromosomal syndromes .Also have autosomal recessive inheritance](https://image.slidesharecdn.com/microcephalyjo-230430183105-ad3aefb0/85/MICROCEPHALY-jo-pptx-9-320.jpg)


















This document defines microcephaly and discusses its classification, etiology, pathogenesis, and diagnosis. Microcephaly is defined as an occipitofrontal circumference more than 3 standard deviations below the mean for age and gender. It can be classified as congenital or postnatal, genetic or environmental, symmetric or asymmetric, and isolated or syndromic. Causes include genetic conditions like primary microcephaly and chromosomal abnormalities, as well as congenital infections, metabolic disorders, and environmental factors. Neuroimaging can aid diagnosis by identifying brain abnormalities and calcifications.
















































































