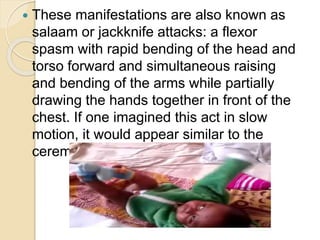
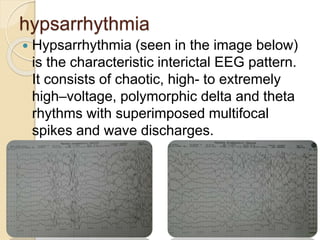
West syndrome is a severe epilepsy syndrome in infants characterized by infantile spasms, a chaotic EEG pattern called hypsarrhythmia, and developmental delays. It was first described in 1841 and is difficult to treat. Treatment options include adrenocorticotropic hormone (ACTH), vigabatrin, prednisone, pyridoxine, and anti-seizure medications like valproic acid. Prognosis depends on the underlying cause but many children experience cognitive impairments or other developmental issues even if seizures are controlled.































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)






