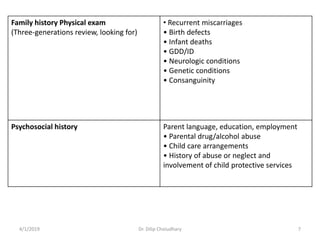
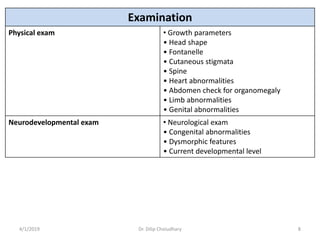
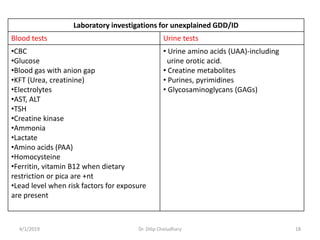
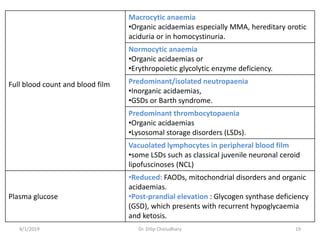
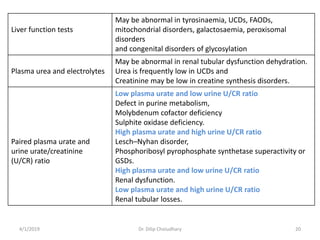
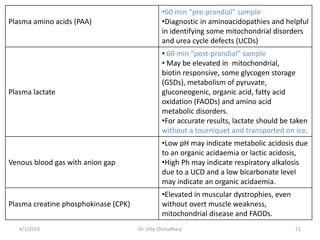
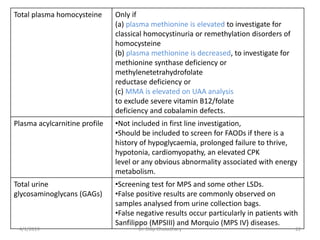
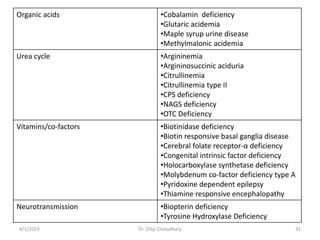
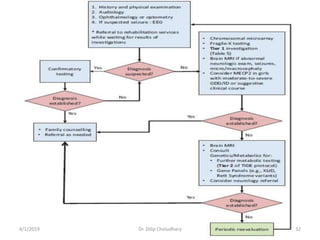
This document provides information on evaluating and investigating children with global developmental delay (GDD) or intellectual disability (ID). It defines GDD and ID and outlines the similar investigations pursued for both, including medical history, physical exam, genetic testing like chromosomal microarray and metabolic testing of blood and urine. The diagnostic yield of investigations is highest when focused on identifying potentially treatable inborn errors of metabolism or single-gene disorders. The document recommends chromosome microarray as the first-line genetic test and discusses other genetic tests, metabolic tests, imaging and neurological evaluation that can help identify underlying etiologies.