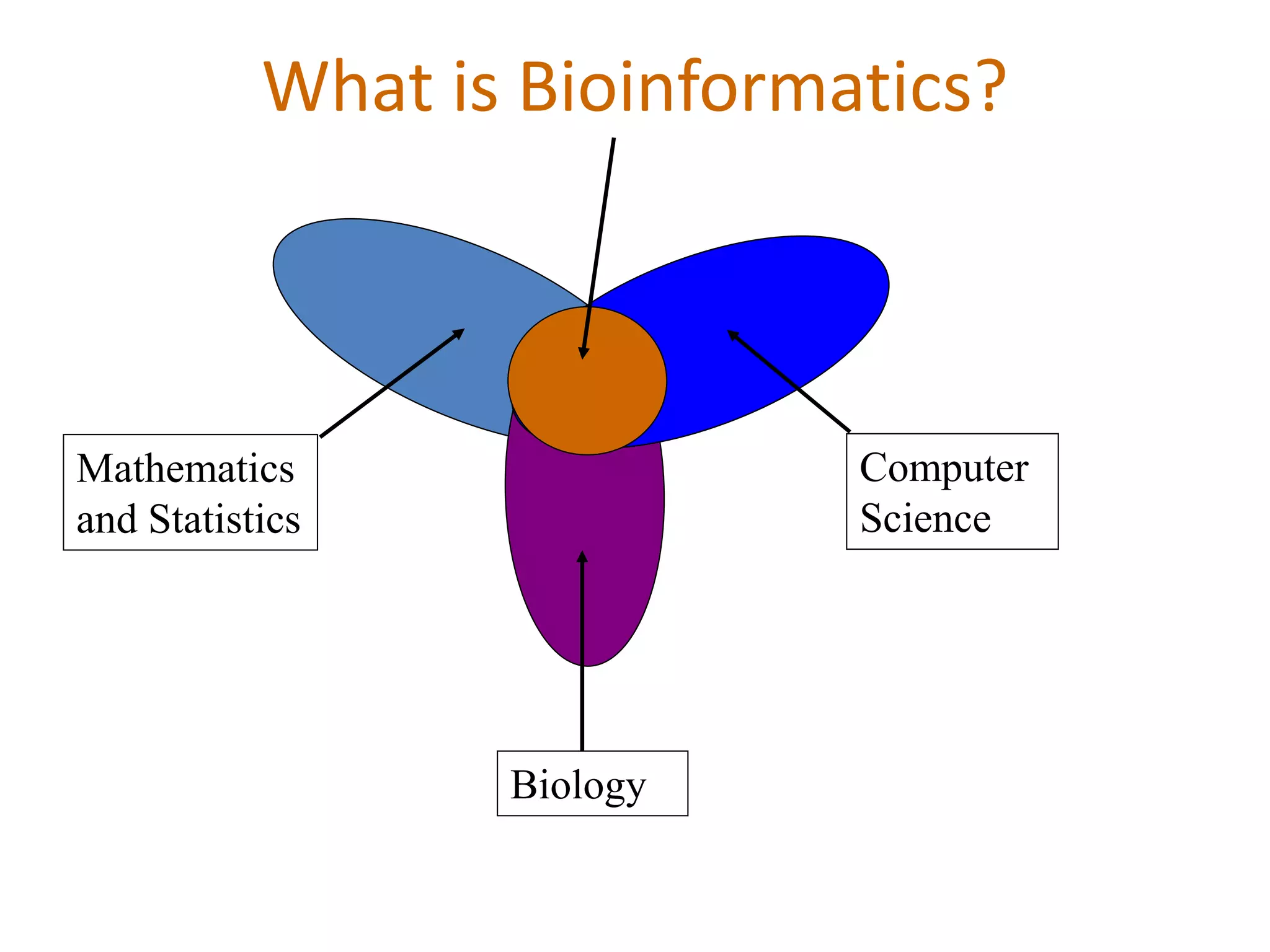
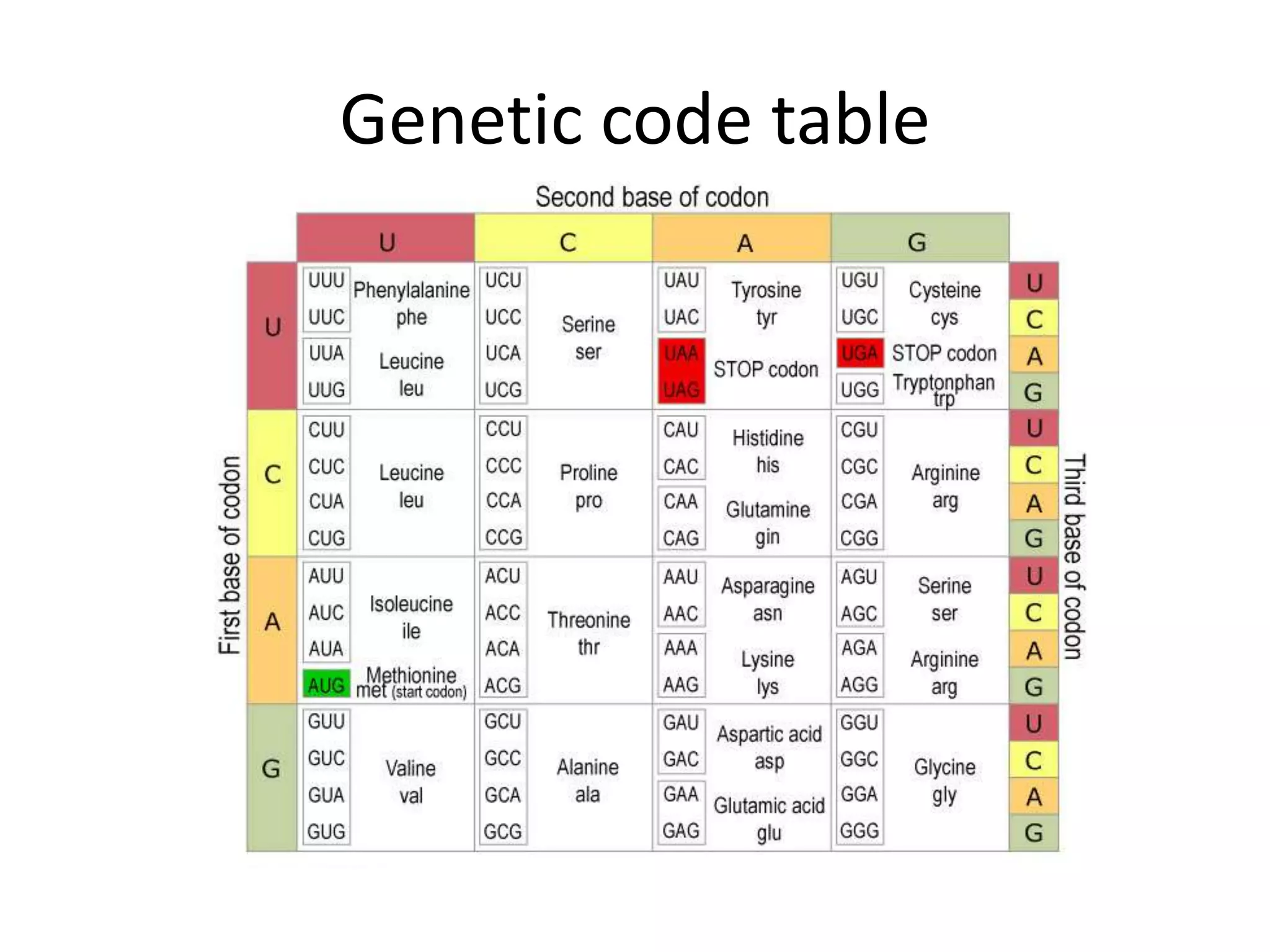
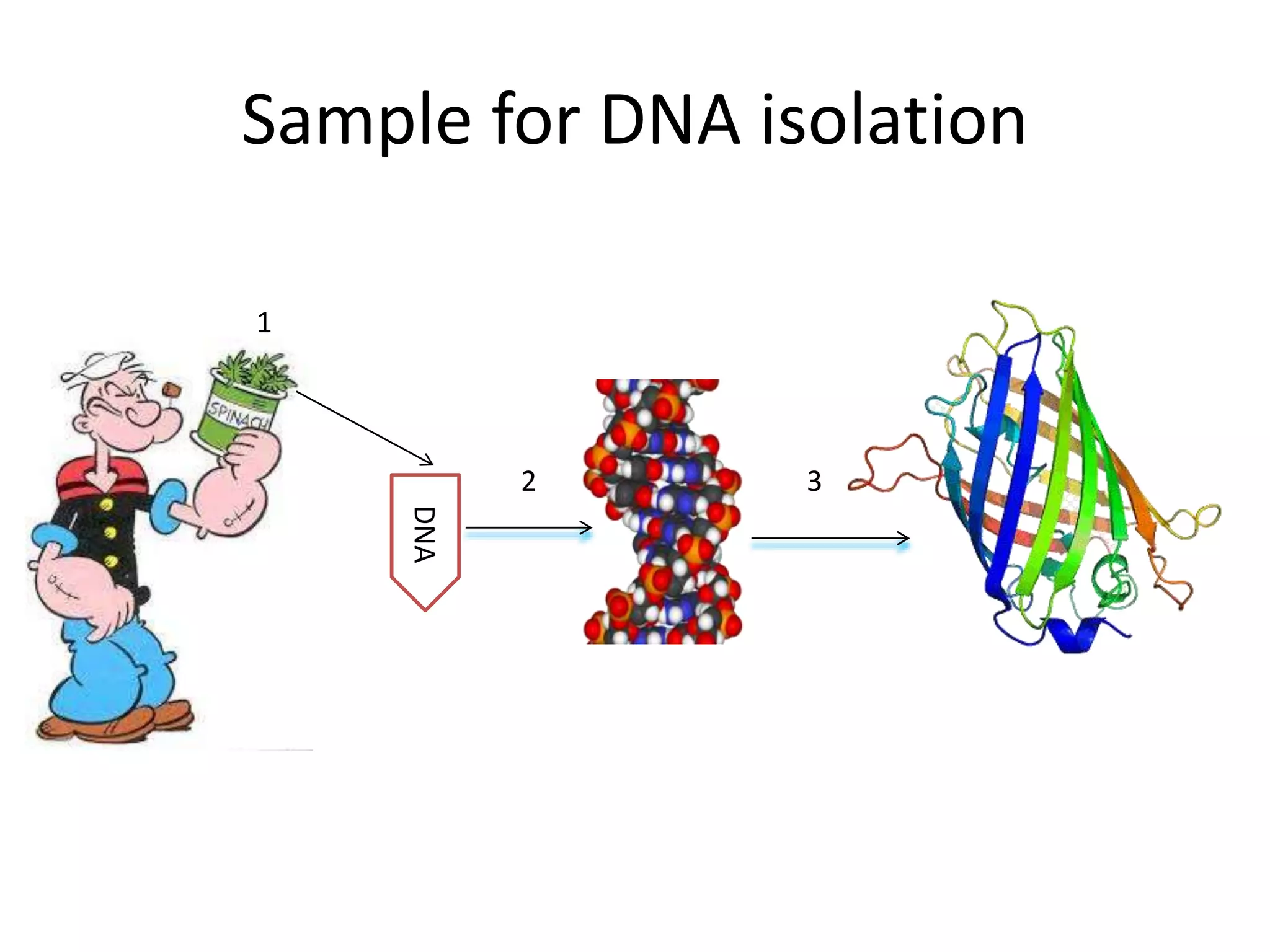
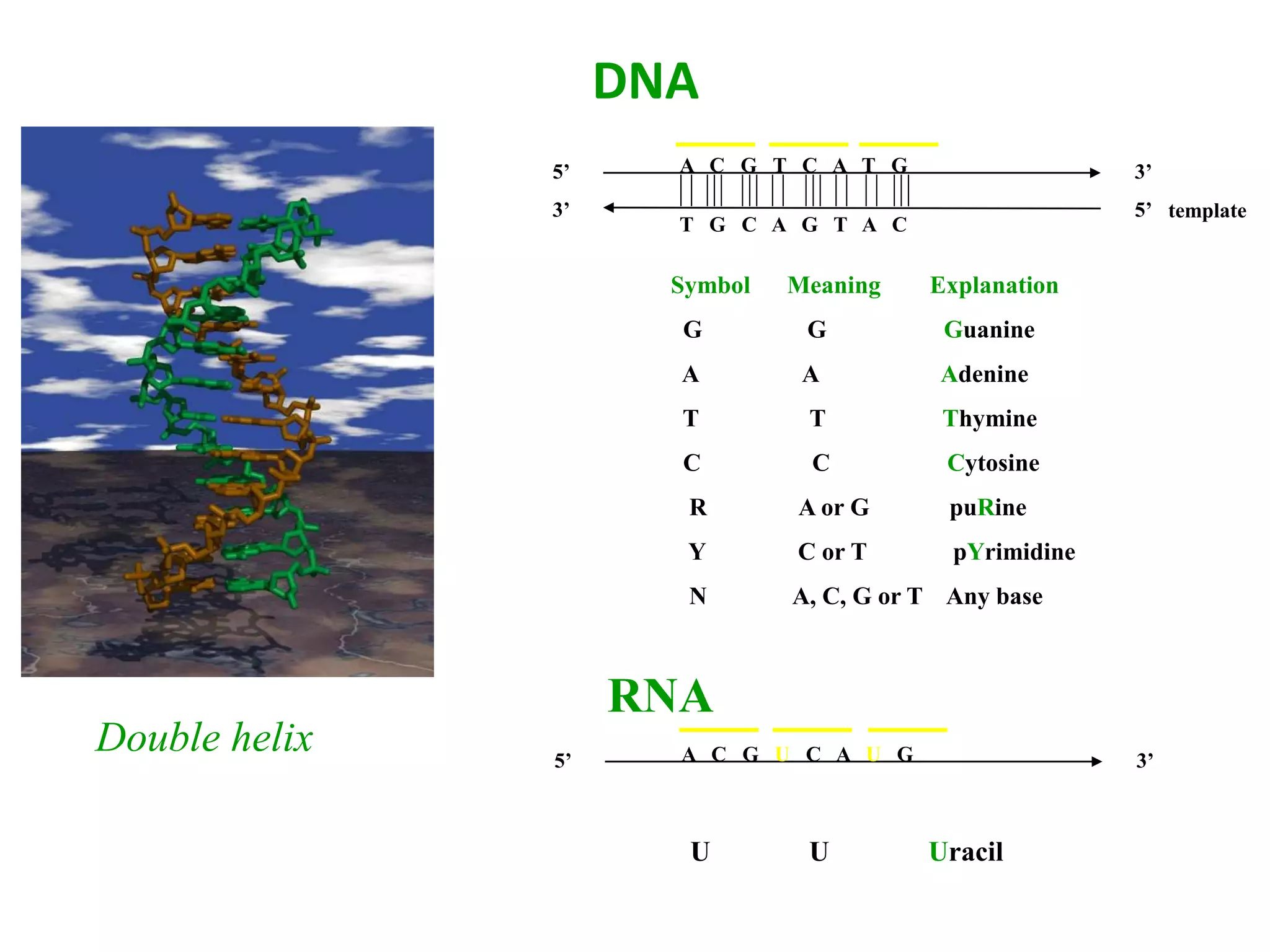
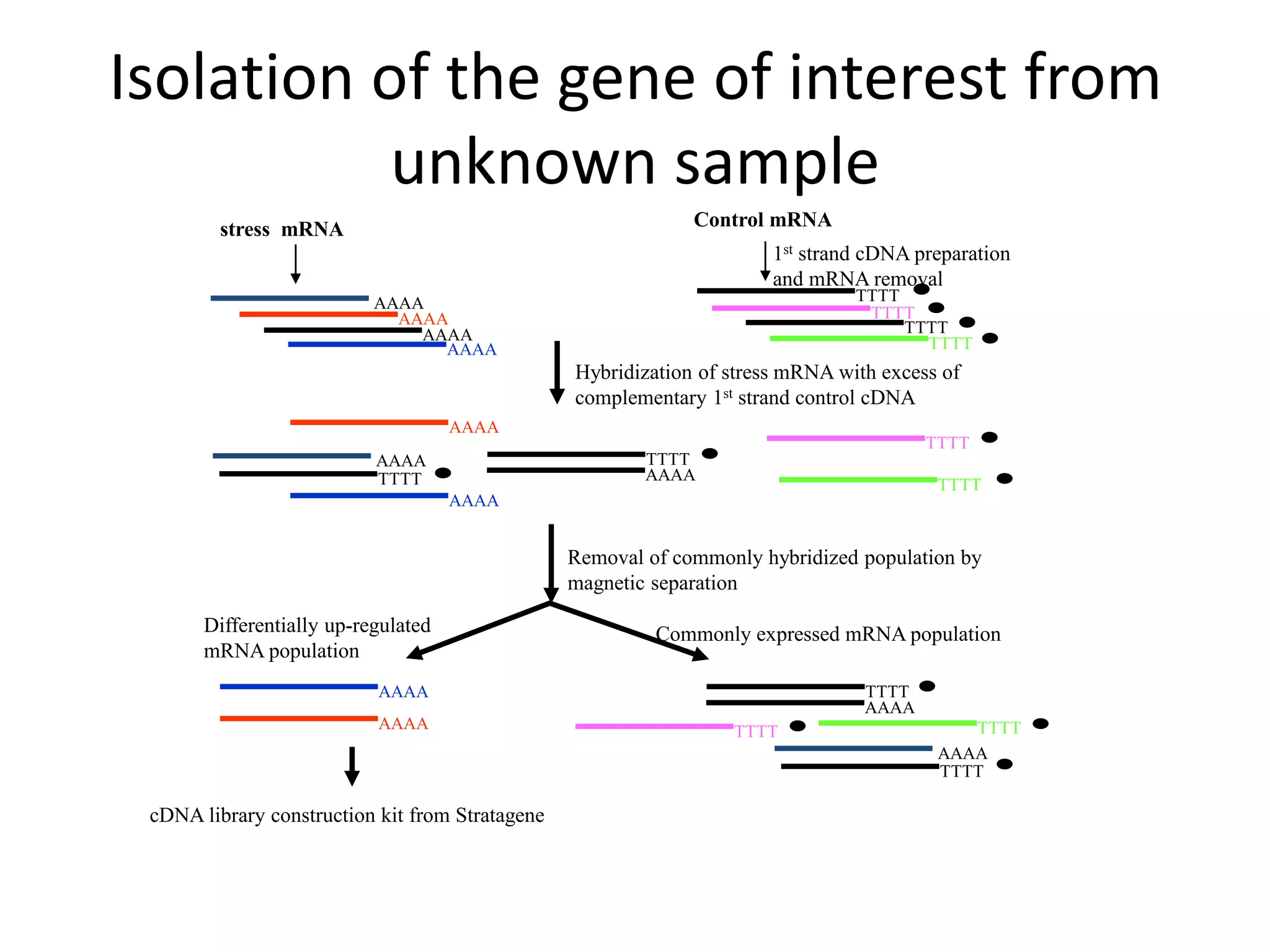
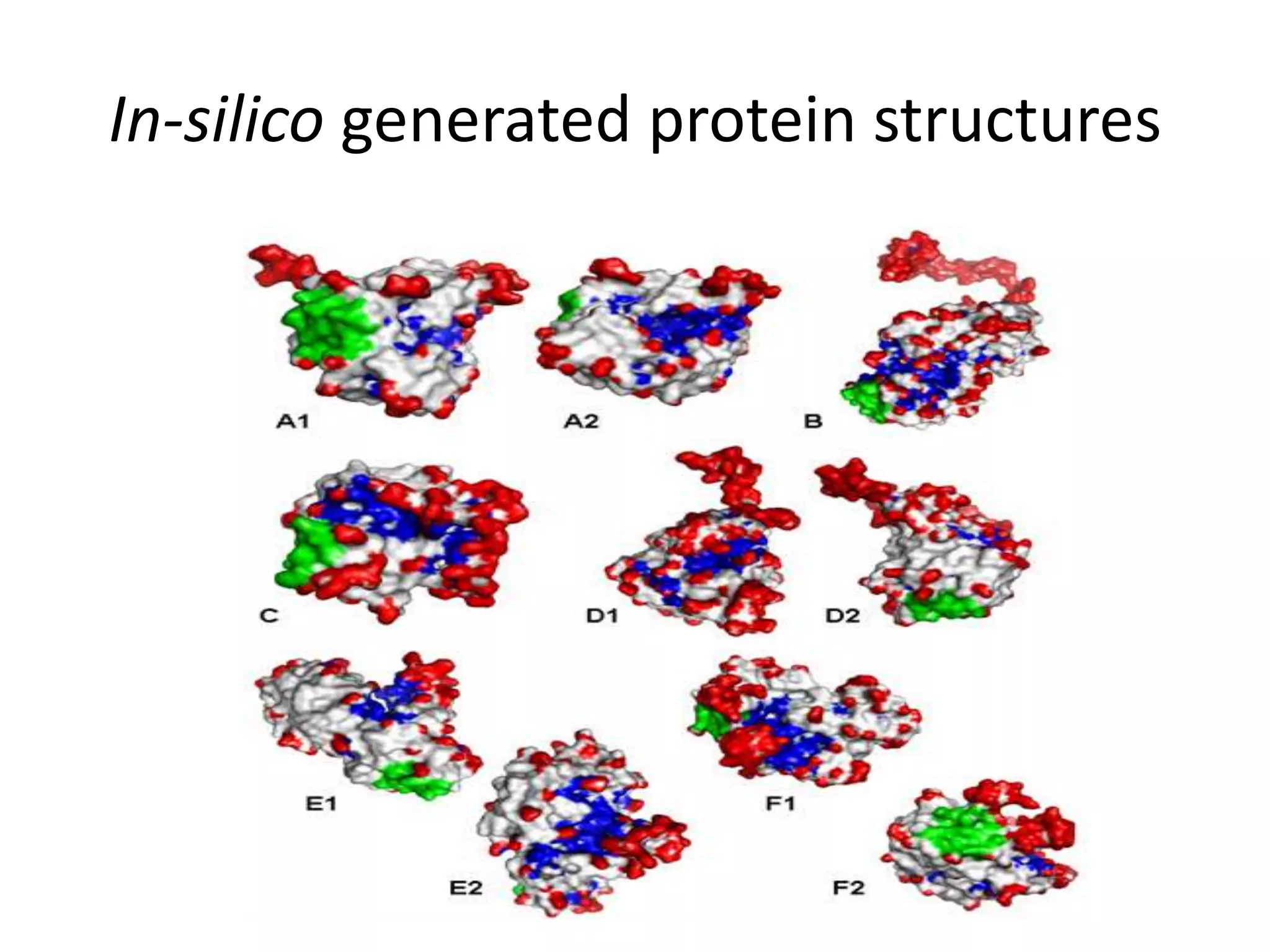
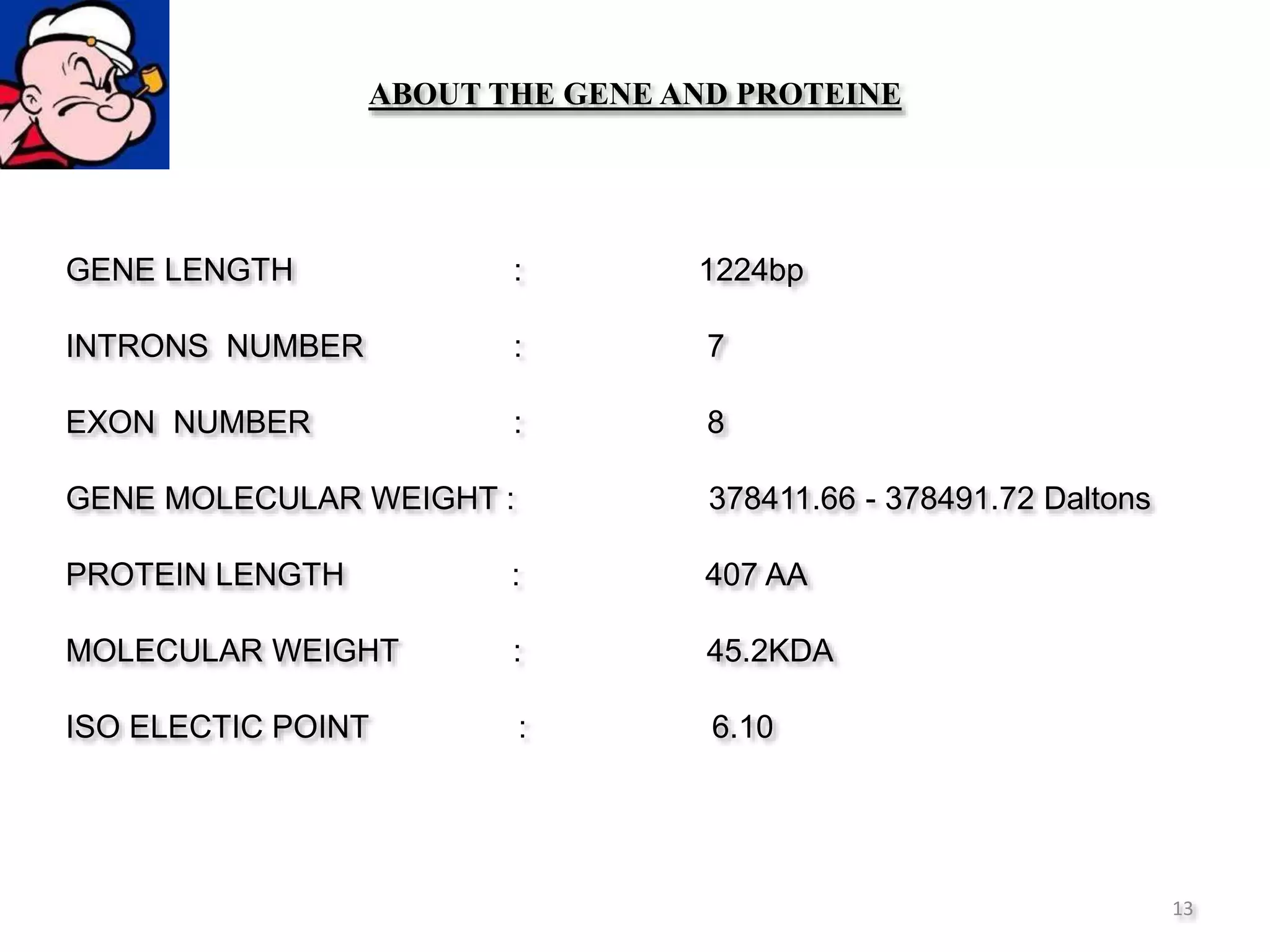
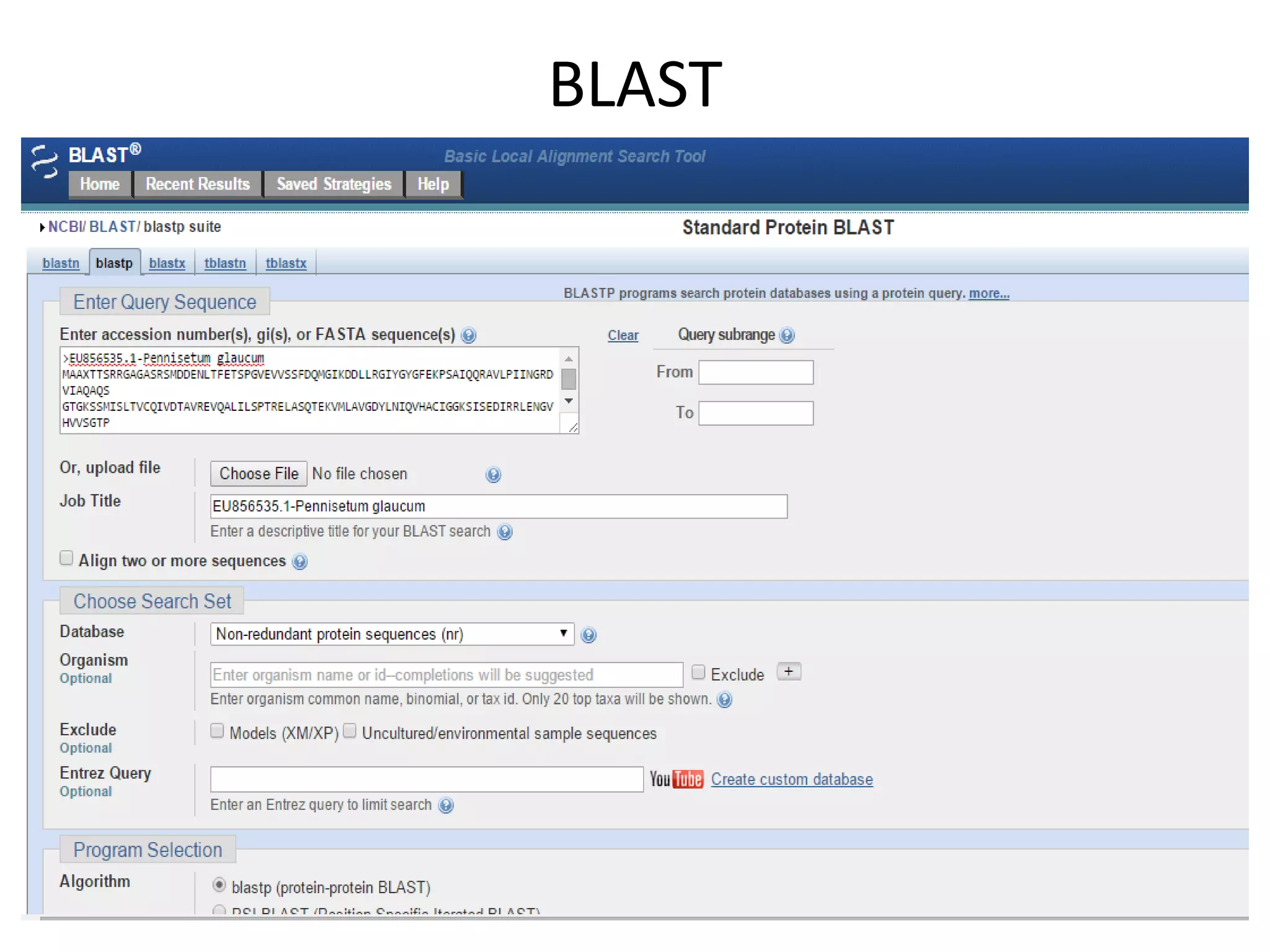
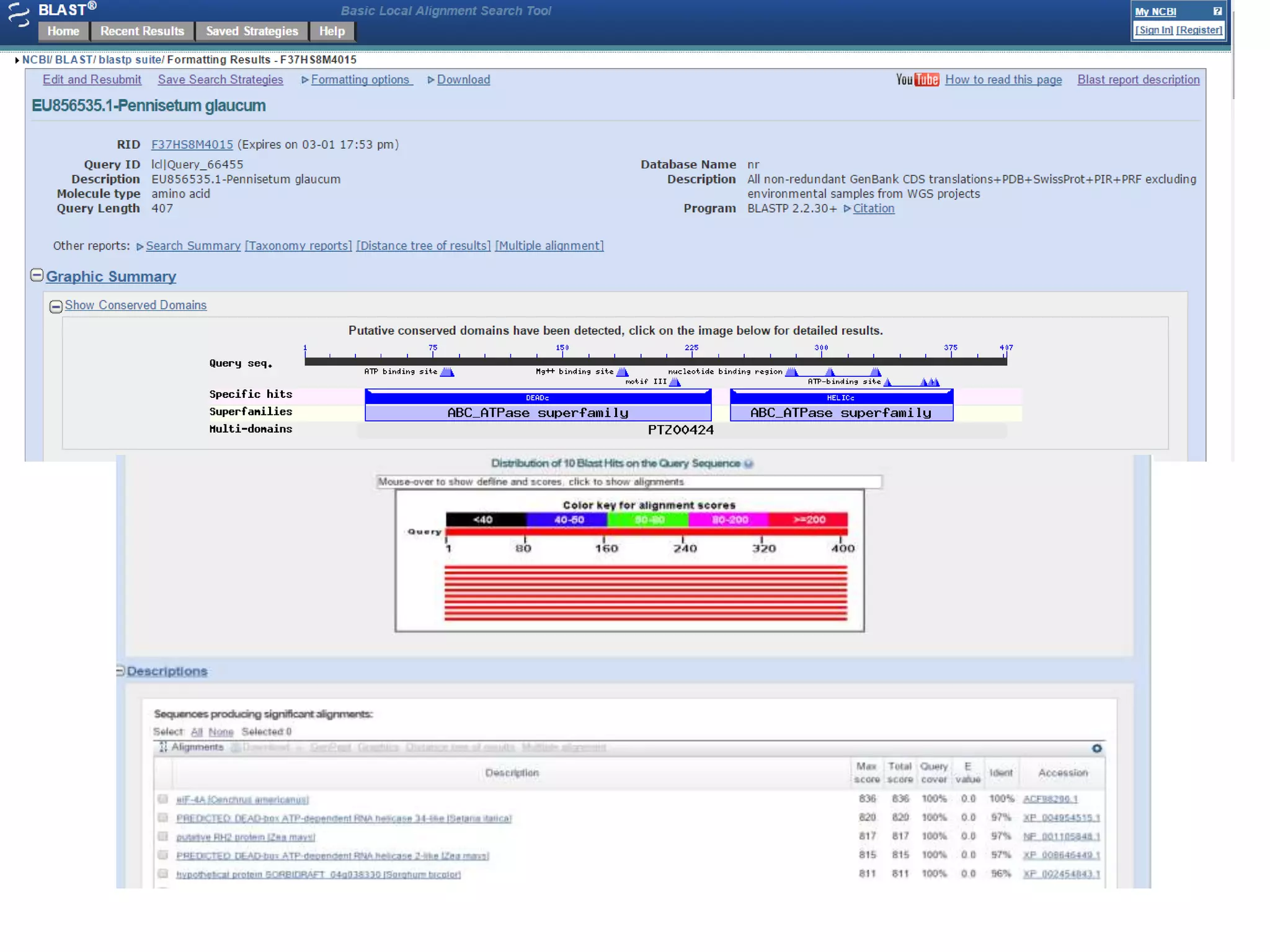
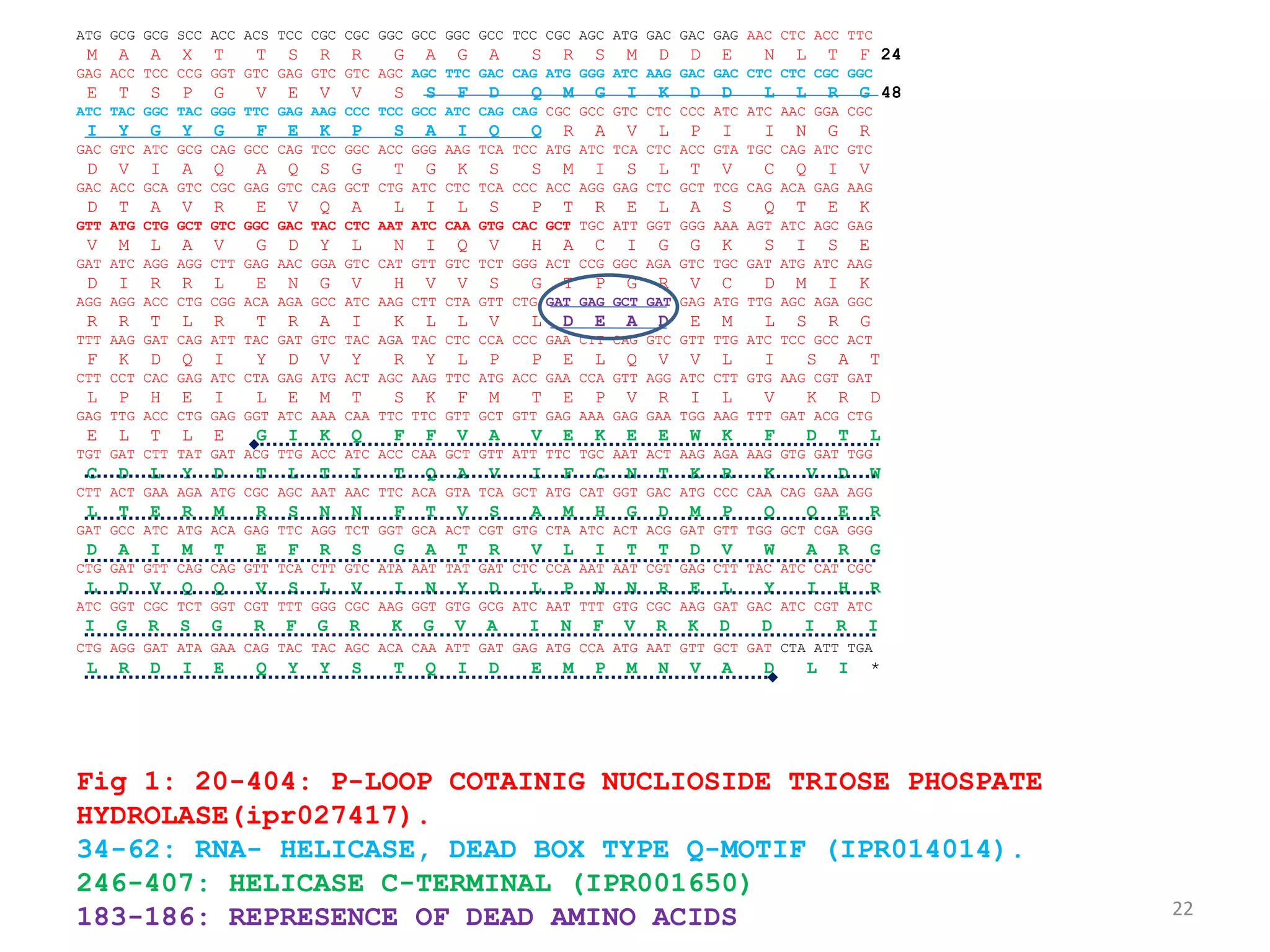
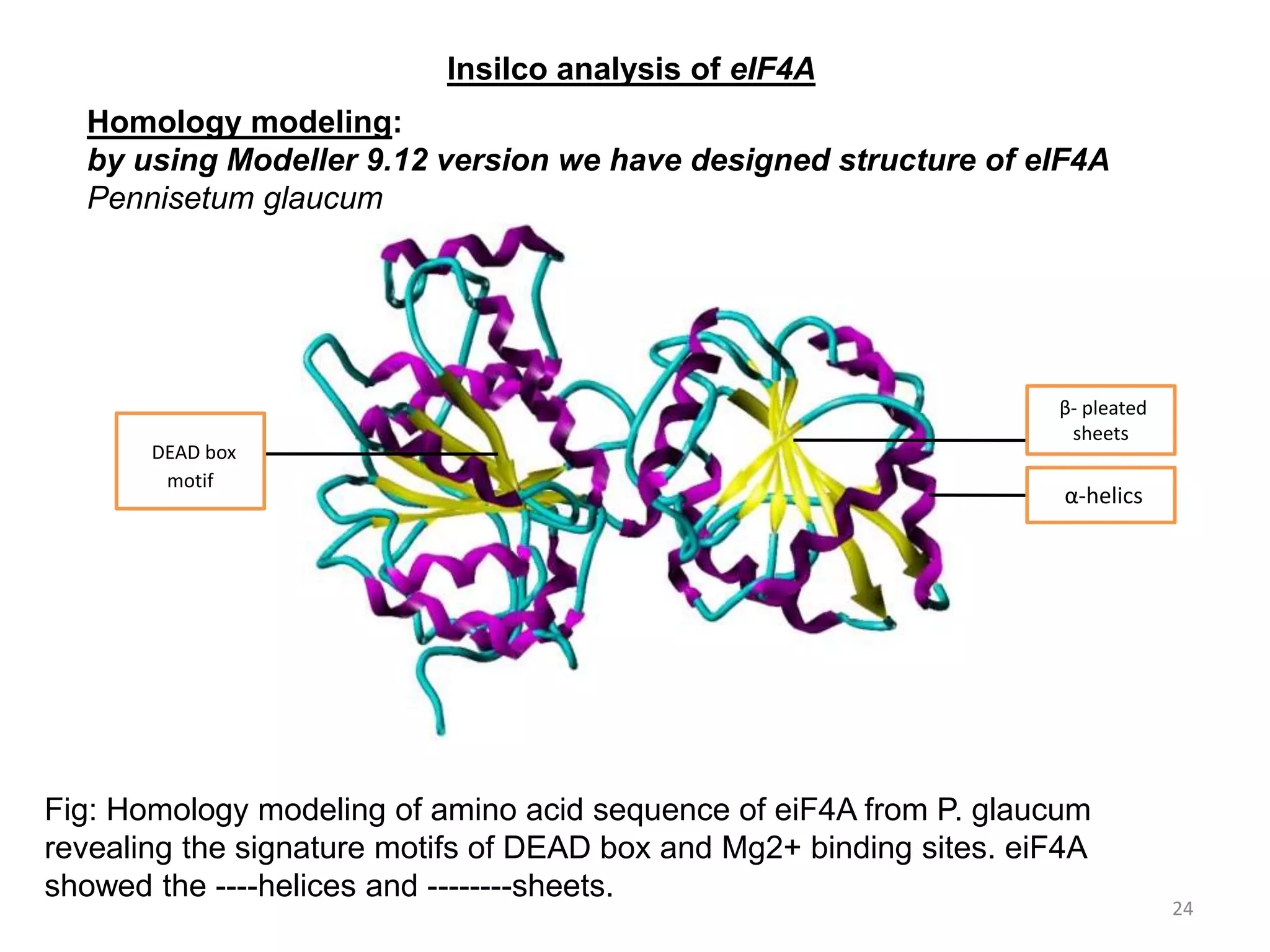
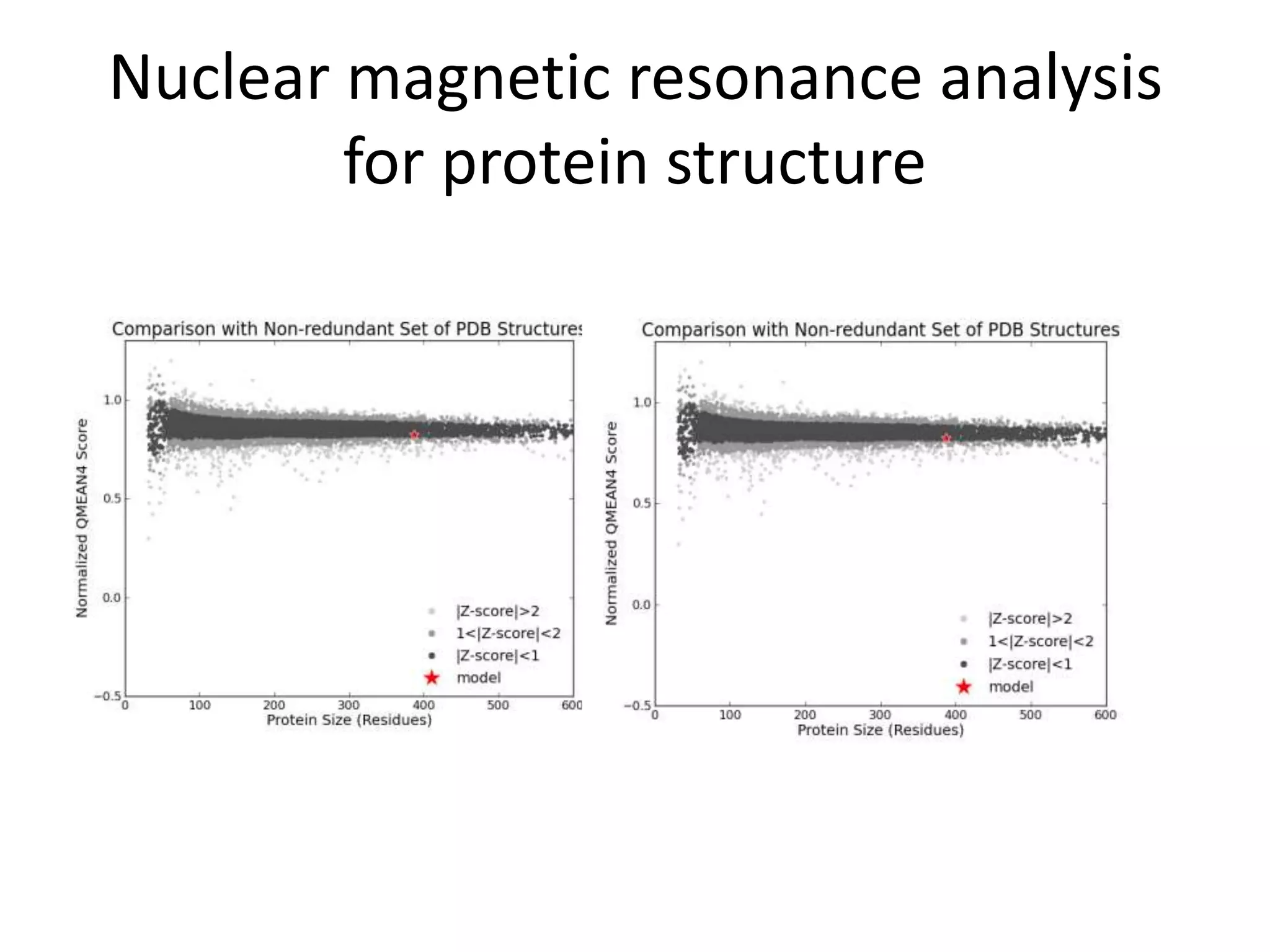
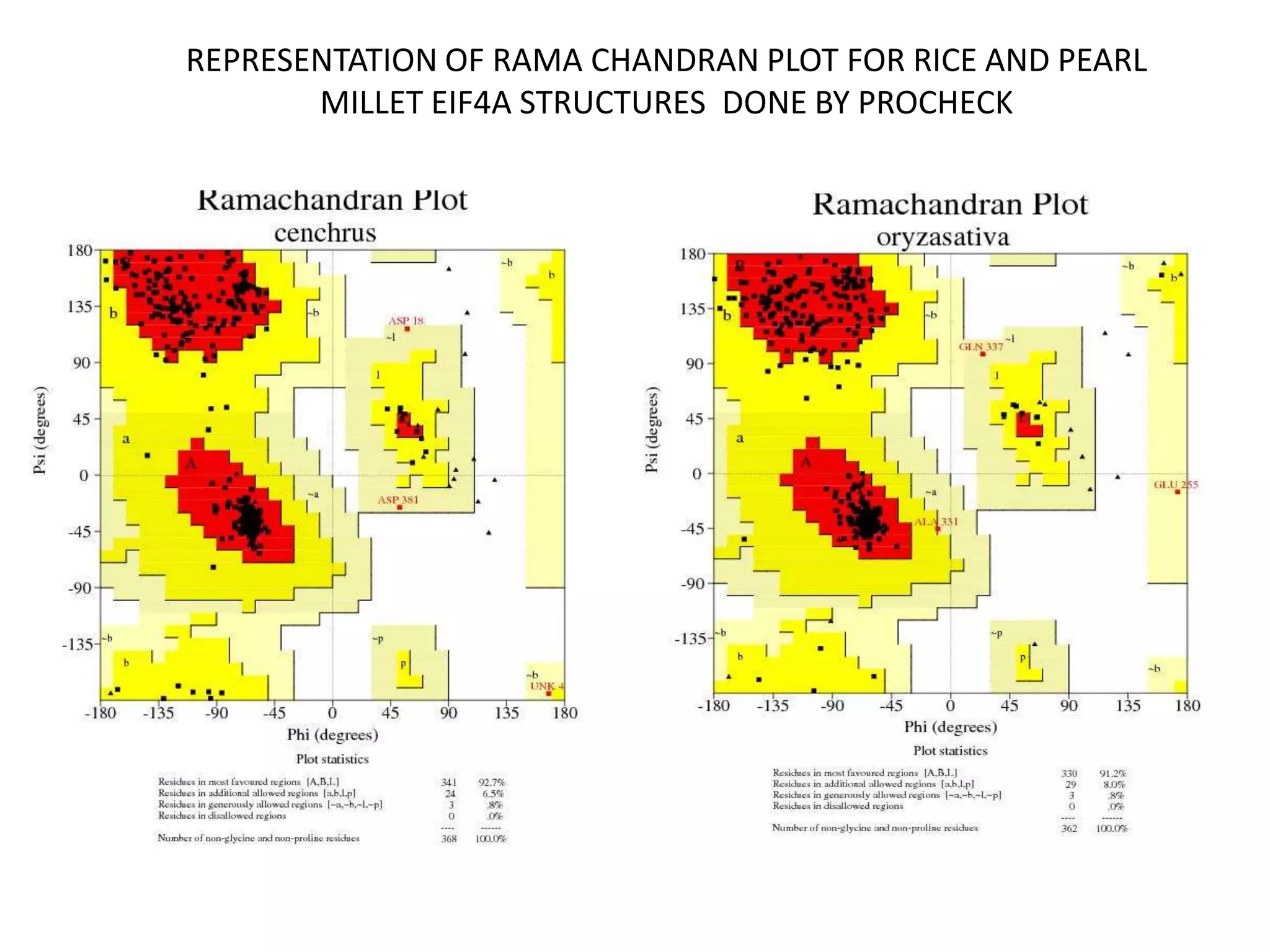
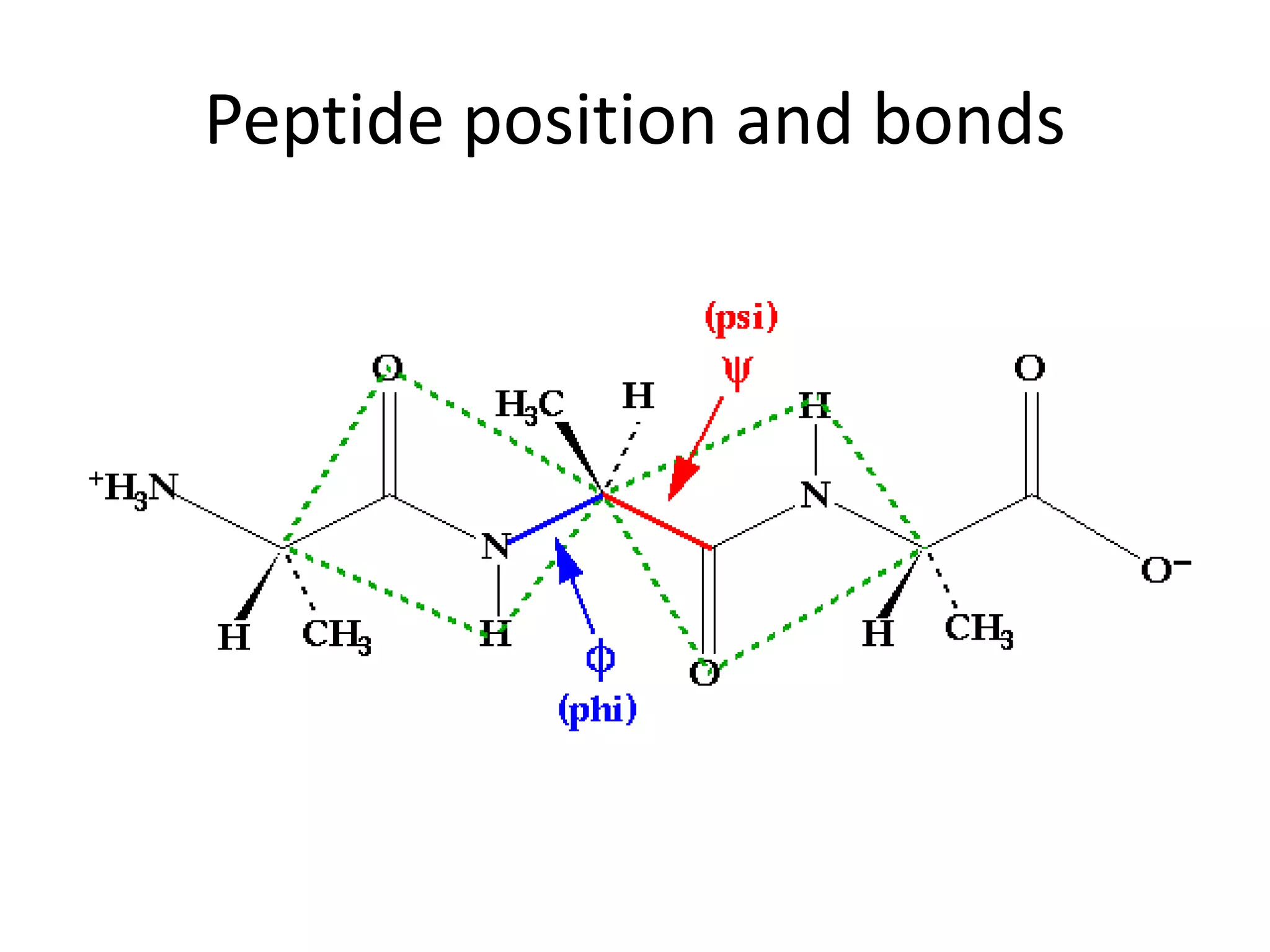
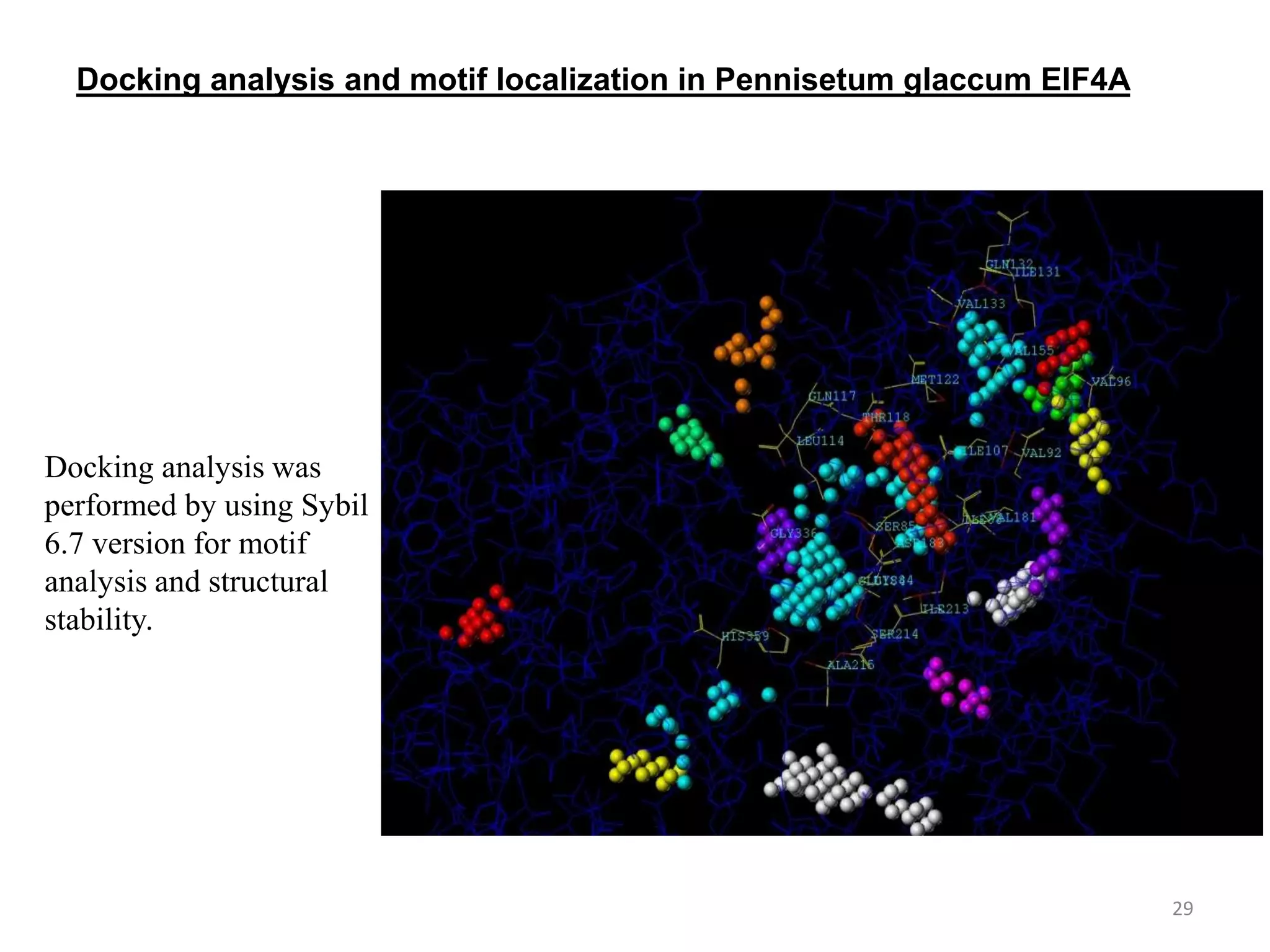
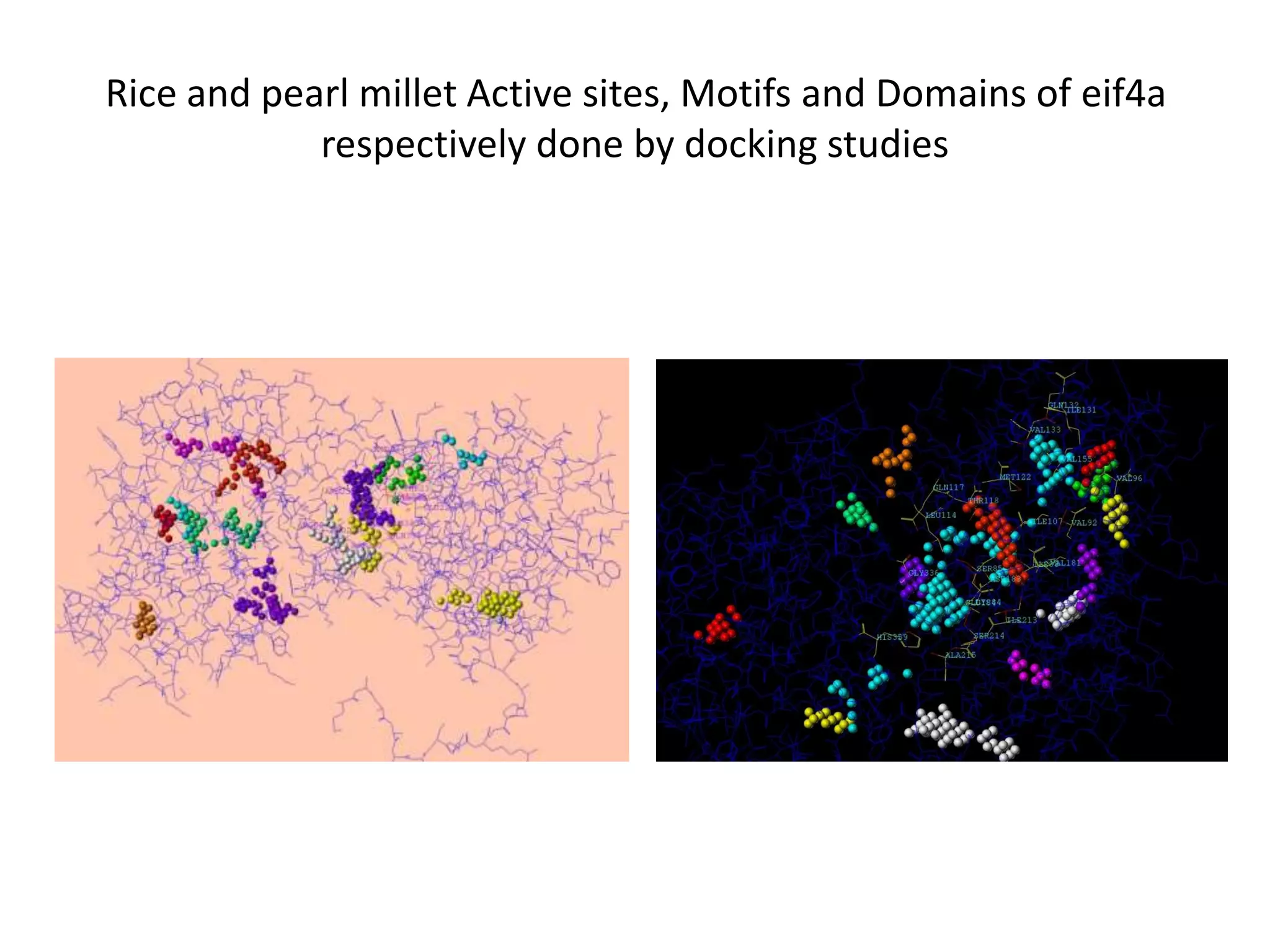
The document discusses in-silico analysis and bioinformatics, focusing on data handling, molecular analysis, and protein structure modeling. It outlines the methodologies involved, including data collection, sequence alignment, phylogenetic analysis, and structural validation techniques for studying gene expression and protein functions. The conclusion emphasizes the accuracy of in-silico studies in guiding research before conducting in-vitro experiments.