Downloaded 13 times






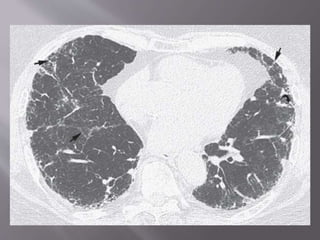

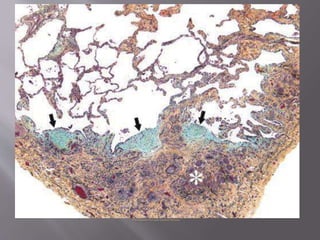


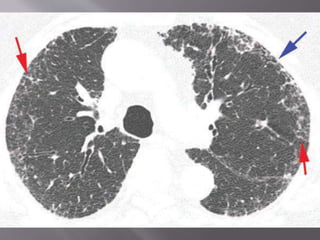

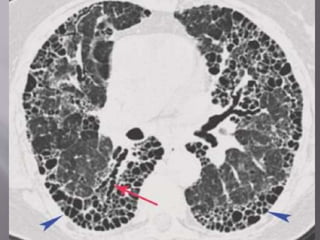

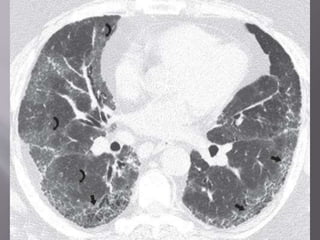



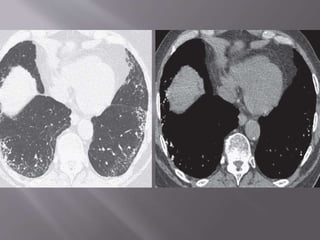

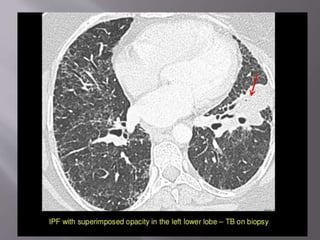


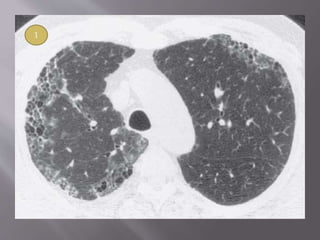
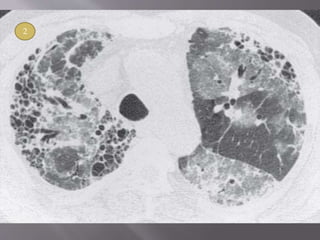



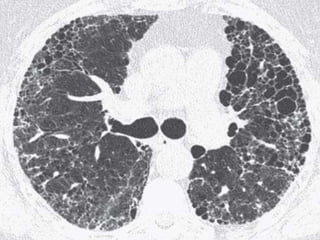
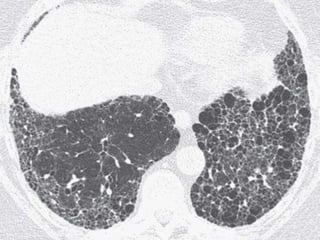


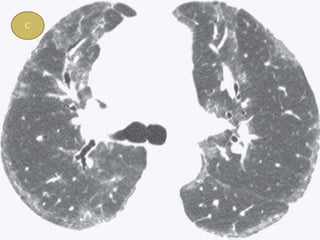

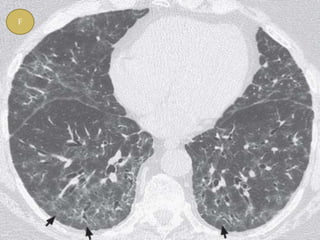

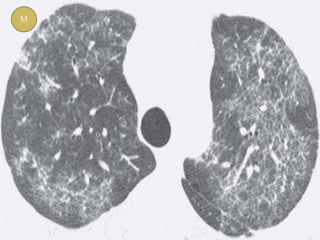

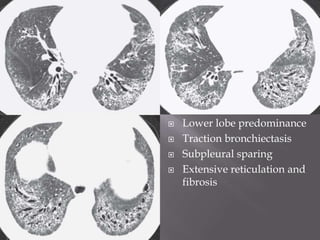

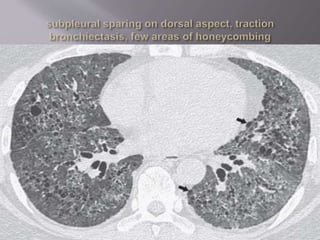
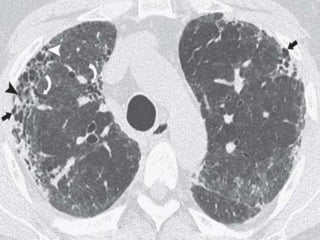



This document discusses non-specific interstitial pneumonia (NSIP), a type of interstitial lung disease. It provides details on: 1. The characteristic CT findings of NSIP including ground glass opacities, irregular linear opacities, and lower lobe predominance with sparing of the subpleural lung. Honeycombing is uncommon. 2. NSIP can be difficult to distinguish from other interstitial lung diseases on CT alone, and a multidisciplinary approach including biopsy may be needed. The extent of honeycombing seen on CT helps distinguish NSIP from idiopathic pulmonary fibrosis. 3. The prognosis of NSIP depends on the subtype, with fibrotic NS















































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)





